Congenital disorder of glycosylation
A congenital disorder of glycosylation (previously called carbohydrate-deficient glycoprotein syndrome) is one of several rare inborn errors of metabolism in which glycosylation of a variety of tissue proteins and/or lipids is deficient or defective. Congenital disorders of glycosylation are sometimes known as CDG syndromes. They often cause serious, sometimes fatal, malfunction of several different organ systems (especially the nervous system, muscles, and intestines) in affected infants.[1] The most common sub-type is PMM2-CDG (formerly known as CDG-Ia) where the genetic defect leads to the loss of phosphomannomutase 2 (PMM2), the enzyme responsible for the conversion of mannose-6-phosphate into mannose-1-phosphate[2]
| Congenital disorders of glycosylation | |
|---|---|
| Specialty | Neurology |
Presentation
Clinical features depend on the molecular pathology of the particular CDG subtype. Common manifestations include ataxia; seizures; retinopathy; liver disease; coagulopathies; failure to thrive (FTT); dysmorphic features (e.g., inverted nipples and subcutaneous fat pads); pericardial effusion, skeletal abnormalities, and hypotonia. If an MRI is obtained, cerebellar hypoplasia is a common finding.[3] Some CDG subtypes, like SSR4-CDG 1y, have been classified as connective tissue disorders.[4]
Ocular abnormalities of PMM2-CDG include: myopia, infantile esotropia, delayed visual maturation, peripheral neuropathy (PN), strabismus, nystagmus, optic disc pallor, and reduced rod function on electroretinography.[5] Three CDG subtypes PMM2-CDG, PMI-CDG, ALG6-CDG can cause congenital hyperinsulinism with hyperinsulinemic hypoglycemia in infancy.[6] Because glycoproteins are involved in many central nervous system processes important during early development, intellectual disability and developmental delays are also common in CDG [7]
N-Glycosylation and known defects
A biologically very important group of carbohydrates is the asparagine (Asn)-linked, or N-linked, oligosaccharides. Their biosynthetic pathway is very complex and involves a hundred or more glycosyltransferases, glycosidases, transporters and synthases. This plethora allows for the formation of a multitude of different final oligosaccharide structures, involved in protein folding, intracellular transport/localization, protein activity, and degradation/half-life. A vast amount of carbohydrate binding molecules (lectins) depend on correct glycosylation for appropriate binding; the selectins, involved in leukocyte extravasation, is a prime example. Their binding depends on a correct fucosylation of cell surface glycoproteins. Lack thereof leads to leukocytosis and increased sensitivity to infections as seen in SLC35C1-CDG(CDG-IIc); caused by a GDP-fucose (Fuc) transporter deficiency.[8] All N-linked oligosaccharides originate from a common lipid-linked oligosaccharide (LLO) precursor, synthesized in the ER on a dolichol-phosphate (Dol-P) anchor. The mature LLO is transferred co-translationally to consensus sequence Asn residues in the nascent protein, and is further modified by trimming and re-building in the Golgi.[9]
Deficiencies in the genes involved in N-linked glycosylation constitute the molecular background of most CDGs.[10]
- Type I defects involve the synthesis and transfer of the LLO
- Type II defects impair the modification process of protein-bound oligosaccharides.
Type I
| Description | Disorder | Product |
|---|---|---|
| The formation of the LLO is initiated by the synthesis of the polyisoprenyl dolichol from farnesyl, a precursor of cholesterol biosynthesis. This step involves at least three genes, DHDDS (encoding dehydrodolichyl diphosphate synthase that is a cis-prenyl transferase), DOLPP1 (a pyrophosphatase) and SRD5A3, encoding a reductase that completes the formation of dolichol. | Recently, exome sequencing showed that mutations in DHDDS cause a disorder with a retinal phenotype (retinitis pigmentosa, a common finding in CDG patients.[11] Further, the intermediary reductase in this process (encoded by SRD5A3), is deficient in SRD5A3-CDG (CDG-Iq).[12] | |
| Dol is then activated to Dol-P via the action of Dol kinase in the ER membrane. | This process is defective in DOLK-CDG (CDG-Im).[13] | 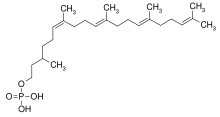 |
| Consecutive N-acetylglucosamine (GlcNAc)- and mannosyltransferases use the nucleotide sugar donors UDP-GlcNAc and GDP-mannose (Man) to form a pyrophosphate-linked seven sugar glycan structure (Man5GlcNAc2-PP-Dol) on the cytoplasmatic side of the ER. | Some of these steps have been found deficient in patients.
|
Man5GlcNAc2-PP-Dol |
| The M5GlcNAc2-structure is then flipped to the ER lumen, via the action of a "flippase" | This is deficient in RFT1-CDG (CDG-In).[18] | |
| Finally, three mannosyltransferases and three glucosyltransferases complete the LLO structure Glc3Man9GlcNAc2-PP-Dol using Dol-P-Man and Dol-P-glucose (Glc) as donors. | There are five known defects:
|
Glc3Man9GlcNAc2-PP-Dol |
| A protein with hitherto unknown activity, MPDU-1, is required for the efficient presentation of Dol-P-Man and Dol-P-Glc. | Its deficiency causes MPDU1-CDG (CDG-If).[24] | |
| The synthesis of GDP-Man is crucial for proper N-glycosylation, as it serves as donor substrate for the formation of Dol-P-Man and the initial Man5GlcNAc2-P-Dol structure. GDP-Man synthesis is linked to glycolysis via the interconversion of fructose-6-P and Man-6-P, catalyzed by phosphomannose isomerase (PMI). | This step is deficient in MPI-CDG (CDG-Ib),[25] which is the only treatable CDG-I subtype. | 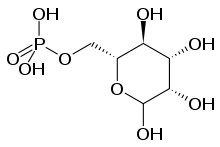 |
| Man-1-P is then formed from Man-6-P, catalyzed by phosphomannomutase (PMM2), and Man-1-P serves as substrate in the GDP-Man synthesis. | Mutations in PMM2 cause PMM2-CDG (CDG-Ia), the most common CDG subtype.[26] | 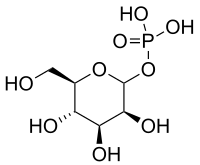 |
| Dol-P-Man is formed via the action of Dol-P-Man synthase, consisting of three subunits; DPM1, DPM2, and DPM3. | Mutations in DPM1 causes DPM1-CDG (CDG-Ie). Mutations in DPM2 (DPM2-CDG) and DPM3 (DPM3-CDG (CDG-Io))[27] cause syndromes with a muscle phenotype resembling an a-dystroglycanopathy, possibly due to lack of Dol-P-Man required for O-mannosylation. | 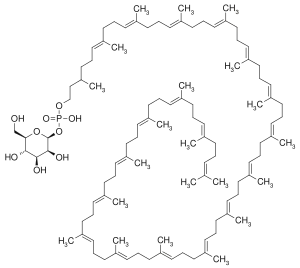 |
| The final Dol-PP-bound 14mer oligosaccharides (Glc3Man9GlcNAc2-PP-Dol) are transferred to consensus Asn residues in the acceptor proteins in the ER lumen, catalyzed by the oligosaccharyltransferase(OST). The OST is composed by several subunits, including DDOST, TUSC3, MAGT1, KRTCAP2 and STT3a and -3b. | Three of these genes have hitherto been shown to be mutated in CDG patients, DDOST (DDOST-CDG (CDG-Ir)), TUSC3 (TUSC3-CDG) and MAGT1 (MAGT1-CDG). |
Type II
The mature LLO chain is next transferred to the growing protein chain, a process catalysed by the oligosaccharyl transferase (OST) complex.[28]
- Once transferred to the protein chain, the oligosaccharide is trimmed by specific glycosidases. This process is vital since the lectin chaperones calnexin and calreticulin, involved in protein quality, bind to the Glc1Man9GlcNAc-structure and assure proper folding. Lack of the first glycosidase (GCS1) causes CDG-IIb.
- Removal of the Glc residues and the first Man residue occurs in the ER.
- The glycoprotein then travels to the Golgi, where a multitude of different structures with different biological activities are formed.
- Mannosidase I creates a Man5GlcNAc2-structure on the protein, but note that this has a different structure than the one made on LLO.
- Next, a GlcNAc residue forms GlcNAc1Man5GlcNAc2, the substrate for a-mannosidase II (aManII).
- aManII then removes two Man residues, creating the substrate for GlcNAc transferase II, which adds a GlcNAc to the second Man branch. This structure serves as substrate for additional galactosylation, fucosylation and sialylation reactions. Additionally, substitution with more GlcNAc residues can yield tri- and tetra-antennary molecules.
Not all structures are fully modified, some remain as high-mannose structures, others as hybrids (one unmodified Man branch and one modified), but the majority become fully modified complex type oligosaccharides.[29]
In addition to glycosidase I, mutations have been found:
- in MGAT2, in GlcNAc transferase II (CDG-IIa)
- in SLC35C1, the GDP-Fuc transporter (CDG-IIc)
- in B4GALT1, a galactosyltransferase (CDG-IId)
- in COG7, the conserved oligomeric Golgi complex-7 (CDG-IIe)
- in SLC35A1, the CMP-sialic acid (NeuAc) transporter (CDG-IIf)
However, since at least 1% of the genome is involved in glycosylation, it is likely that many more defects remain to be found.[30]
Diagnosis
Classification
Historically, CDGs are classified as Types I and II (CDG-I and CDG-II), depending on the nature and location of the biochemical defect in the metabolic pathway relative to the action of oligosaccharyltransferase. The most commonly used screening method for CDG, analysis of transferrin glycosylation status by isoelectric focusing, ESI-MS, or other techniques, distinguish between these subtypes in so called Type I and Type II patterns.[31]
Currently, over 130 subtypes of CDG have been described [32].[7]
Since 2009, most researchers use a different nomenclature based on the gene defect (e.g. CDG-Ia = PMM2-CDG, CDG-Ib = PMI-CDG, CDG-Ic = ALG6-CDG etc.).[33] The reason for the new nomenclature was the fact that proteins not directly involved in glycan synthesis (such as members of the COG-family[34] and vesicular H+-ATPase)[35] were found to be causing the glycosylation defect in some CDG patients.
Also, defects disturbing other glycosylation pathways than the N-linked one are included in this classification. Examples are the α-dystroglycanopathies (e.g. POMT1/POMT2-CDG (Walker-Warburg syndrome and Muscle-Eye-Brain syndrome)) with deficiencies in O-mannosylation of proteins; O-xylosylglycan synthesis defects (EXT1/EXT2-CDG (hereditary multiple exostoses) and B4GALT7-CDG (Ehlers-Danlos syndrome, progeroid variant)); O-fucosylglycan synthesis (B3GALTL-CDG (Peter's plus syndrome) and LFNG-CDG (spondylocostal dysostosis III)).[36]
Type I
- Type I disorders involve disrupted synthesis of the lipid-linked oligosaccharide precursor (LLO) or its transfer to the protein.
Types include:
| Type | OMIM | Gene | Locus |
|---|---|---|---|
| Ia (PMM2-CDG) | 212065 | PMM2 | 16p13.3-p13.2 |
| Ib (MPI-CDG) | 602579 | MPI | 15q22-qter |
| Ic (ALG6-CDG) | 603147 | ALG6 | 1p22.3 |
| Id (ALG3-CDG) | 601110 | ALG3 | 3q27 |
| Ie (DPM1-CDG) | 608799 | DPM1 | 20q13.13 |
| If (MPDU1-CDG) | 609180 | MPDU1 | 17p13.1-p12 |
| Ig (ALG12-CDG) | 607143 | ALG12 | 22q13.33 |
| Ih (ALG8-CDG) | 608104 | ALG8 | 11pter-p15.5 |
| Ii (ALG2-CDG) | 607906 | ALG2 | 9q22 |
| Ij (DPAGT1-CDG) | 608093 | DPAGT1 | 11q23.3 |
| Ik (ALG1-CDG) | 608540 | ALG1 | 16p13.3 |
| 1L (ALG9-CDG) | 608776 | ALG9 | 11q23 |
| Im (DOLK-CDG) | 610768 | DOLK | 9q34.11 |
| In (RFT1-CDG) | 612015 | RFT1 | 3p21.1 |
| Io (DPM3-CDG) | 612937 | DPM3 | 1q12-q21 |
| Ip (ALG11-CDG) | 613661 | ALG11 | 13q14.3 |
| Iq (SRD5A3-CDG) | 612379 | SRD5A3 | 4q12 |
| Ir (DDOST-CDG) | 614507 | DDOST | 1p36.12 |
| It (PGM1-CDG)
(formerly GSD-XIV) |
614921 | PGM1 | 1p31.3 |
| DPM2-CDG | n/a | DPM2 | 9q34.13 |
| TUSC3-CDG | 611093 | TUSC3 | 8p22 |
| MAGT1-CDG | 300716 | MAGT1 | X21.1 |
| DHDDS-CDG | 613861 | DHDDS | 1p36.11 |
| I/IIx | 212067 | n/a | n/a |
Type II
- Type II disorders involve malfunctioning trimming/processing of the protein-bound oligosaccharide chain.
Types include:
| Type | OMIM | Gene | Locus |
|---|---|---|---|
| IIa (MGAT2-CDG) | 212066 | MGAT2 | 14q21 |
| IIb (GCS1-CDG) | 606056 | GCS1 | 2p13-p12 |
| IIc (SLC335C1-CDG; Leukocyte adhesion deficiency II)) | 266265 | SLC35C1 | 11p11.2 |
| IId (B4GALT1-CDG) | 607091 | B4GALT1 | 9p13 |
| IIe (COG7-CDG) | 608779 | COG7 | 16p |
| IIf (SLC35A1-CDG) | 603585 | SLC35A1 | 6q15 |
| IIg (COG1-CDG) | 611209 | COG1 | 17q25.1 |
| IIh (COG8-CDG) | 611182 | COG8 | 16q22.1 |
| IIi (COG5-CDG) | 613612 | COG5 | 7q31 |
| IIj (COG4-CDG) | 613489 | COG4 | 16q22.1 |
| IIL (COG6-CDG) | n/a | COG6 | 13q14.11 |
| IIT (CDG2T) | 618885 | GALNT2 | |
| ATP6V0A2-CDG (autosomal recessive cutis laxa type 2a (ARCL-2A)) | 219200 | ATP6V0A2 | 12q24.31 |
| MAN1B1-CDG (Mental retardation, autosomal recessive 15) | 614202 | MAN1B1 | 9q34.3 |
| ST3GAL3-CDG (Mental retardation, autosomal recessive 12) | 611090 | ST3GAL3 | 1p34.1 |
Disorders of O-mannosylation
- Disorders with deficient α-dystroglycan O-mannosylation.
Mutations in several genes have been associated with the traditional clinical syndromes, termed muscular dystrophy-dystroglycanopathies (MDDG). A new nomenclature based on clinical severity and genetic cause was recently proposed by OMIM.[37] The severity classifications are A (severe), B (intermediate), and C (mild). The subtypes are numbered one to six according to the genetic cause, in the following order: (1) POMT1, (2) POMT2, (3) POMGNT1, (4) FKTN, (5) FKRP, and (6) LARGE.[38]
Most common severe types include:
| Name | OMIM | Gene | Locus |
|---|---|---|---|
| POMT1-CDG (MDDGA1;Walker-Warburg syndrome) | 236670 | POMT1 | 9q34.13 |
| POMT2-CDG (MDDGA2;Walker-Warburg syndrome) | 613150 | POMT2 | 14q24.3 |
| POMGNT1-CDG (MDDGA3; muscle-eye-brain) | 253280 | POMGNT1 | 1p34.1 |
| FKTN-CDG (MDDGA4; Fukuyama congenital muscular dystrophy) | 253800 | FKTN | 9q31.2 |
| FKRP-CDG (MDDGB5; MDC1C) | 606612 | FKRP | 19q13.32 |
| LARGE-CDG (MDDGB6; MDC1D) | 608840 | LARGE | 22q12.3 |
Treatment
No treatment is available for most of these disorders. Mannose supplementation relieves the symptoms in MPI-CDG for the most part,[39] even though the hepatic fibrosis may persist.[40] Fucose supplementation has had a partial effect on some SLC35C1-CDG patients.[41]
History
The first CDG patients (twin sisters) were described in 1980 by Jaeken et al.[42] Their main features were psychomotor retardation, cerebral and cerebellar atrophy and fluctuating hormone levels (e.g.prolactin, FSH and GH). During the next 15 years the underlying defect remained unknown but since the plasmaprotein transferrin was underglycosylated (as shown by e.g. isoelectric focusing), the new syndrome was named carbohydrate-deficient glycoprotein syndrome (CDGS)[1] Its "classic" phenotype included psychomotor retardation, ataxia, strabismus, anomalies (fat pads and inverted nipples) and coagulopathy.
In 1994, a new phenotype was described and named CDGS-II.[43] In 1995, Van Schaftingen and Jaeken showed that CDGS-I (now PMM2-CDG) was caused by the deficiency of the enzyme phosphomannomutase. This enzyme is responsible for the interconversion of mannose-6-phosphate and mannose-1-phosphate, and its deficiency leads to a shortage in GDP-mannose and dolichol (Dol)-mannose (Man), two donors required for the synthesis of the lipid-linked oligosaccharide precursor of N-linked glycosylation.[44]
In 1998, Niehues described a new CDG syndrome, MPI-CDG, which is caused by mutations in the enzyme metabolically upstream of PMM2, phosphomannose isomerase (PMI).[25] A functional therapy for MPI-CDG, alimentary mannose was also described.[25]
The characterization of new defects took increased and several new Type I and Type II defects were delineated.[45]
In 2012, Need described the first case of a congenital disorder of deglycosylation, NGLY1 deficiency.[46] A 2014 study of NGLY1 deficient patients found similarities with traditional congenital disorders of glycosylation.[47]
References
- Jaeken J, Carchon H (1993). "The carbohydrate-deficient glycoprotein syndromes: an overview". Journal of Inherited Metabolic Disease. 16 (5): 813–20. doi:10.1007/bf00714272. PMID 8295395. S2CID 10219089.
- Schollen, E.; Pardon, E.; Heykants, L.; Renard, J.; Doggett, N.A.; Callen, D.F.; Cassiman, J.J.; Mathijs, G. (1998). "Comparative analysis of the phosphomannomutase genes PMM1, PMM2 and PMM2psi: the sequence variation in the processed pseudogene is a reflection of the mutations found in the functional gene". Human Molecular Genetics. 7 (2): 157–164. doi:10.1093/hmg/7.2.157. PMID 9425221.
- Paprocka, J.; Jezela-Stanek, A.; Tylki-Szyma´nska, A.; Grunewald, S. (2021). "Congenital Disorders of Glycosylation from a Neurological Perspective". Brain Sciences. 11 (88): 88. doi:10.3390/brainsci11010088. PMC 7827962. PMID 33440761.
- Castiglioni, C.; Feillet, F.; Barnerias, C.; Wiedemann, A.; Muchart, J.; Cortes, F.; Hernando-Davalillo, C.; Montero, R.; Dupré, T.; Bruneel, N.; Seta, N.; Vuillaumier-Barrot, S.; Serrano, M. (2021). "Expanding the phenotype of X-linked SSR4-CDG: Connective tissue implications". Human Mutation. 42 (2): 142–149. doi:10.1002/humu.24151. PMID 33300232. S2CID 228087106.
- Jensen H, Kjaergaard S, Klie F, Moller HU (June 2003). "Ophthalmic manifestations of congenital disorder of glycosylation type 1a". Ophthalmic Genetics. 24 (2): 81–8. doi:10.1076/opge.24.2.81.13994. PMID 12789572. S2CID 29341185.
- Sun L, Eklund EA, Chung WK, Wang C, Cohen J, Freeze HH (July 2005). "Congenital disorder of glycosylation is presenting with hyperinsulinemic hypoglycemia and islet cell hyperplasia". The Journal of Clinical Endocrinology and Metabolism. 90 (7): 4371–5. doi:10.1210/jc.2005-0250. PMID 15840742.
- Freeze HH, Eklund EA, Ng BG, Patterson MC (May 2012). "Neurology of inherited glycosylation disorders". The Lancet. Neurology. 11 (5): 453–66. doi:10.1016/S1474-4422(12)70040-6. PMC 3625645. PMID 22516080.
- Marquardt, T.; Brune, T.; Luhn, K.; Zimmer, K.; Korner, C.; Fabritz, L.; van der Werft, N.; Vormoor, J.; Freeze, H.H.; Louwen, F.; Bierman, B.; Harms, E.; von Figura, K.; Vestweber, D.; Koch, H.G. (1999). "Leukocyte adhesion deficiency II syndrome, a generalized defect in fucose metabolism". The Journal of Pediatrics. 134 (6): 681–688. doi:10.1016/S0022-3476(99)70281-7. PMC 7095022. PMID 10356134.
- Breitling, J.; Aebi, M. (2013). "N-Linked Protein Glycosylation in the Endoplasmic Reticulum". Cold Spring Harbor Perspectives in Biology. 5 (8): a013359. doi:10.1101/cshperspect.a013359. PMC 3721281. PMID 23751184.
- Jaeken, Jaak (2016). "Glycosylation and its Disorders: General Overview☆". Reference Module in Biomedical Sciences. doi:10.1016/B978-0-12-801238-3.04632-8. ISBN 9780128012383.
- Züchner S, Dallman J, Wen R, Beecham G, Naj A, Farooq A, Kohli MA, Whitehead PL, Hulme W, Konidari I, Edwards YJ, Cai G, Peter I, Seo D, Buxbaum JD, Haines JL, Blanton S, Young J, Alfonso E, Vance JM, Lam BL, Peričak-Vance MA (February 2011). "Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa". American Journal of Human Genetics. 88 (2): 201–6. doi:10.1016/j.ajhg.2011.01.001. PMC 3035708. PMID 21295283.
- Cantagrel V, Lefeber DJ, Ng BG, Guan Z, Silhavy JL, Bielas SL, Lehle L, Hombauer H, Adamowicz M, Swiezewska E, De Brouwer AP, Blümel P, Sykut-Cegielska J, Houliston S, Swistun D, Ali BR, Dobyns WB, Babovic-Vuksanovic D, van Bokhoven H, Wevers RA, Raetz CR, Freeze HH, Morava E, Al-Gazali L, Gleeson JG (2010). "SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder". Cell. 142 (2): 203–17. doi:10.1016/j.cell.2010.06.001. PMC 2940322. PMID 20637498.
- Kranz C, Jungeblut C, Denecke J, Erlekotte A, Sohlbach C, Debus V, Kehl HG, Harms E, Reith A, Reichel S, Grobe H, Hammersen G, Schwarzer U, Marquardt T (March 2007). "A defect in dolichol phosphate biosynthesis causes a new inherited disorder with death in early infancy". American Journal of Human Genetics. 80 (3): 433–40. doi:10.1086/512130. PMC 1821118. PMID 17273964.
- Wu X, Rush JS, Karaoglu D, Krasnewich D, Lubinsky MS, Waechter CJ, Gilmore R, Freeze HH (August 2003). "Deficiency of UDP-GlcNAc:Dolichol Phosphate N-Acetylglucosamine-1 Phosphate Transferase (DPAGT1) causes a novel congenital disorder of Glycosylation Type Ij". Human Mutation. 22 (2): 144–50. doi:10.1002/humu.10239. PMID 12872255. S2CID 35331823.
- Grubenmann CE, Frank CG, Hülsmeier AJ, Schollen E, Matthijs G, Mayatepek E, Berger EG, Aebi M, Hennet T (March 2004). "Deficiency of the first mannosylation step in the N-glycosylation pathway causes congenital disorder of glycosylation type Ik". Human Molecular Genetics. 13 (5): 535–42. doi:10.1093/hmg/ddh050. PMID 14709599.
- Thiel C, Schwarz M, Peng J, Grzmil M, Hasilik M, Braulke T, Kohlschütter A, von Figura K, Lehle L, Körner C (June 2003). "A new type of congenital disorders of glycosylation (CDG-Ii) provides new insights into the early steps of dolichol-linked oligosaccharide biosynthesis". The Journal of Biological Chemistry. 278 (25): 22498–505. doi:10.1074/jbc.m302850200. PMID 12684507.
- Rind N, Schmeiser V, Thiel C, Absmanner B, Lübbehusen J, Hocks J, Apeshiotis N, Wilichowski E, Lehle L, Körner C (April 2010). "A severe human metabolic disease caused by deficiency of the endoplasmatic mannosyltransferase hALG11 leads to congenital disorder of glycosylation-Ip". Human Molecular Genetics. 19 (8): 1413–24. doi:10.1093/hmg/ddq016. PMID 20080937.
- Vleugels W, Haeuptle MA, Ng BG, Michalski JC, Battini R, Dionisi-Vici C, Ludman MD, Jaeken J, Foulquier F, Freeze HH, Matthijs G, Hennet T (October 2009). "RFT1 deficiency in three novel CDG patients". Human Mutation. 30 (10): 1428–34. doi:10.1002/humu.21085. PMC 3869400. PMID 19701946.
- Körner C, Knauer R, Stephani U, Marquardt T, Lehle L, von Figura K (December 1999). "Carbohydrate deficient glycoprotein syndrome type IV: deficiency of dolichyl-P-Man:Man(5)GlcNAc(2)-PP-dolichyl mannosyltransferase". The EMBO Journal. 18 (23): 6816–22. doi:10.1093/emboj/18.23.6816. PMC 1171744. PMID 10581255.
- Frank CG, Grubenmann CE, Eyaid W, Berger EG, Aebi M, Hennet T (July 2004). "Identification and functional analysis of a defect in the human ALG9 gene: definition of congenital disorder of glycosylation type IL". American Journal of Human Genetics. 75 (1): 146–50. doi:10.1086/422367. PMC 1181998. PMID 15148656.
- Chantret I, Dupré T, Delenda C, Bucher S, Dancourt J, Barnier A, Charollais A, Heron D, Bader-Meunier B, Danos O, Seta N, Durand G, Oriol R, Codogno P, Moore SE (July 2002). "Congenital disorders of glycosylation type Ig is defined by a deficiency in dolichyl-P-mannose:Man7GlcNAc2-PP-dolichyl mannosyltransferase". The Journal of Biological Chemistry. 277 (28): 25815–22. doi:10.1074/jbc.m203285200. PMID 11983712.
- Körner C, Knauer R, Holzbach U, Hanefeld F, Lehle L, von Figura K (1998). "Carbohydrate-deficient glycoprotein syndrome type V: deficiency of dolichyl-P-Glc:Man9GlcNAc2-PP-dolichyl glucosyltransferase". Proceedings of the National Academy of Sciences of the United States of America. 95 (22): 13200–5. Bibcode:1998PNAS...9513200K. doi:10.1073/pnas.95.22.13200. PMC 23759. PMID 9789065.
- Chantret I, Dancourt J, Dupré T, Delenda C, Bucher S, Vuillaumier-Barrot S, Ogier de Baulny H, Peletan C, Danos O, Seta N, Durand G, Oriol R, Codogno P, Moore SE (March 2003). "A deficiency in dolichyl-P-glucose:Glc1Man9GlcNAc2-PP-dolichyl alpha3-glucosyltransferase defines a new subtype of congenital disorders of glycosylation". The Journal of Biological Chemistry. 278 (11): 9962–71. doi:10.1074/jbc.m211950200. PMID 12480927.
- Kranz C, Denecke J, Lehrman MA, Ray S, Kienz P, Kreissel G, Sagi D, Peter-Katalinic J, Freeze HH, Schmid T, Jackowski-Dohrmann S, Harms E, Marquardt T (2001). "A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG-If)". The Journal of Clinical Investigation. 108 (11): 1613–9. doi:10.1172/JCI13635. PMC 200991. PMID 11733556.
- Niehues R, Hasilik M, Alton G, Körner C, Schiebe-Sukumar M, Koch HG, Zimmer KP, Wu R, Harms E, Reiter K, von Figura K, Freeze HH, Harms HK, Marquardt T (1998). "Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy". The Journal of Clinical Investigation. 101 (7): 1414–20. doi:10.1172/JCI2350. PMC 508719. PMID 9525984.
- Matthijs G, Schollen E, Pardon E, Veiga-Da-Cunha M, Jaeken J, Cassiman JJ, Van Schaftingen E (May 1997). "Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome)". Nature Genetics. 16 (1): 88–92. doi:10.1038/ng0597-88. PMID 9140401. S2CID 22959423.
- Lefeber DJ, Schönberger J, Morava E, Guillard M, Huyben KM, Verrijp K, Grafakou O, Evangeliou A, Preijers FW, Manta P, Yildiz J, Grünewald S, Spilioti M, van den Elzen C, Klein D, Hess D, Ashida H, Hofsteenge J, Maeda Y, van den Heuvel L, Lammens M, Lehle L, Wevers RA (July 2009). "Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies". American Journal of Human Genetics. 85 (1): 76–86. doi:10.1016/j.ajhg.2009.06.006. PMC 2706967. PMID 19576565.
- Freeze, H.H. (2013). "Understanding human glycosylation disorders: biochemistry leads the charge". The Journal of Biological Chemistry. 288 (10): 6936–6945. doi:10.1074/jbc.R112.429274. PMC 3591604. PMID 23329837.
- Schachter, H. (1984). "Glycoproteins: their structure, biosynthesis and possible clinical implications". Clinical Biochemistry. 17 (1): 3–14. doi:10.1016/S0009-9120(84)90360-6. PMID 6368044.
- Jaeken, J. (2013). Congenital Disorders of Glycosylation from a Neurological Perspective. Handbook of Clinical Neurology. Vol. 113. pp. 1737–43. doi:10.1016/B978-0-444-59565-2.00044-7. PMID 23622397.
- Freeze, H.H. (2007). "Congenital Disorders of Glycosylation: CDG-I, CDG-II, and beyond". Current Molecular Medicine. 4 (7): 389–96. doi:10.2174/156652407780831548. PMID 17584079.
- Ondruskova, N.; Cechova, A.; Hansikova, H.; Honzik, T.; Jaeken, J. (2020). "Congenital Disorders of Glycosylation: Still "hot" in 2020". General Subjects. 1865 (2021): 129751. doi:10.1016/j.bbagen.2020.129751. PMID 32991969. S2CID 222159507.
- Jaeken, J., Hennet, T., Matthijs, G., and Freeze, H.H. (2009) CDG nomenclature: time for a change! Biochim Biophys Acta. 1792, 825-6.
- Wu, X., Steet, R.A., Bohorov, O., Bakker, J., Newell, J., Krieger, M., Spaapen, L., Kornfeld, S., and Freeze, H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. (2004) Nat. Med. 10, 518-23.
- Kornak U, Reynders E, Dimopoulou A, van Reeuwijk J, Fischer B, Rajab A, Budde B, Nürnberg P, Foulquier F, Lefeber D, Urban Z, Gruenewald S, Annaert W, Brunner HG, van Bokhoven H, Wevers R, Morava E, Matthijs G, Van Maldergem L, Mundlos S (January 2008). "Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2". Nature Genetics. 40 (1): 32–4. doi:10.1038/ng.2007.45. PMID 18157129. S2CID 23318808.
- Yang, A.; Ng, B.; Moore, S.A.; Rush, J.; Waechter, C.J.; Raymond, K.M.; Willer, T.; Campbell, K.P.; Freeze, H.H.; Mehta, L. (2013). "Congenital disorder of glycosylation due to DPM1 mutations presenting with dystroglycanopathy-type congenital muscular dystrophy". Mol Genet Metab. 110 (3): 345–351. doi:10.1016/j.ymgme.2013.06.016. PMC 3800268. PMID 23856421.
- Amberger J, Bocchini C, Hamosh A (May 2011). "A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®)". Human Mutation. 32 (5): 564–7. doi:10.1002/humu.21466. PMID 21472891.
- Jaeken, J.; Hennet, T.; Mtthijs, G.; Freeze, H.H. (2009). "CDG Nomenclature: Time for a Change". Biochim Biophys Acta. 1792 (9): 825–826. doi:10.1016/j.bbadis.2009.08.005. PMC 3917312. PMID 19765534.
- Mention K, Lacaille F, Valayannopoulos V, Romano S, Kuster A, Cretz M, Zaidan H, Galmiche L, Jaubert F, de Keyzer Y, Seta N, de Lonlay P (2008). "Development of liver disease despite mannose treatment in two patients with CDG-Ib". Molecular Genetics and Metabolism. 93 (1): 40–3. doi:10.1016/j.ymgme.2007.08.126. PMID 17945525.
- Westphal V, Kjaergaard S, Davis JA, Peterson SM, Skovby F, Freeze HH (2001). "Genetic and metabolic analysis of the first adult with congenital disorder of glycosylation type Ib: long-term outcome and effects of mannose supplementation". Molecular Genetics and Metabolism. 73 (1): 77–85. doi:10.1006/mgme.2001.3161. PMID 11350186.
- Eklund EA, Freeze HH (2006). "The congenital disorders of glycosylation: a multifaceted group of syndromes". NeuroRx. 3 (2): 254–63. doi:10.1016/j.nurx.2006.01.012. PMC 3593443. PMID 16554263.
- Jaeken, J., Vanderschueren-Lodeweyckx, M., Casaer, P., Snoeck, L., Corbeel, L., Eggermont, E., and Eeckels, R. (1980) Pediatr Res 14, 179
- Jaeken J, Schachter H, Carchon H, De Cock P, Coddeville B, Spik G (1994). "Carbohydrate deficient glycoprotein syndrome type II: a deficiency in Golgi localised N-acetyl-glucosaminyltransferase II". Archives of Disease in Childhood. 71 (2): 123–7. doi:10.1136/adc.71.2.123. PMC 1029941. PMID 7944531.
- Schaftingen, E.V.; Jaeken, J. (1995). "Phosphomannomutase deficiency is a cause of carbohydrate-deficient glycoprotein syndrome type I". FEBS Letters. 377 (3): 318–320. doi:10.1016/0014-5793(95)01357-1. PMID 8549746. S2CID 321749.
- Haeuptle MA, Hennet T (2009). "Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides" (PDF). Human Mutation. 30 (12): 1628–41. doi:10.1002/humu.21126. PMID 19862844. S2CID 46281092.
- Need AC, Shashi V, Hitomi Y, Schoch K, Shianna KV, McDonald MT, Meisler MH, Goldstein DB (June 2012). "Clinical application of exome sequencing in undiagnosed genetic conditions". Journal of Medical Genetics. 49 (6): 353–61. doi:10.1136/jmedgenet-2012-100819. PMC 3375064. PMID 22581936.
- Enns GM, Shashi V, Bainbridge M, Gambello MJ, Zahir FR, Bast T, et al. (October 2014). "Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway". Genetics in Medicine. 16 (10): 751–8. doi:10.1038/gim.2014.22. PMC 4243708. PMID 24651605.
External links
- GeneReviews/NIH/NCBI/UW entry on PMM2-CDG (CDG-Ia)Carbohydrate-Deficient Glycoprotein Syndrome, Type 1a; Congenital Disorder of Glycosylation Type 1a; Jaeken Syndrome
- OMIM entries on Carbohydrate-Deficient Glycoprotein Syndrome, Type 1a; Congenital Disorder of Glycosylation Type 1a; Jaeken Syndrome
- GeneReviews/NIH/NCBI/UW entry on Congenital Disorders of Glycosylation Overview