Hemolytic–uremic syndrome
Hemolytic–uremic syndrome (HUS) is a group of blood disorders characterized by low red blood cells, acute kidney failure, and low platelets.[1][3] Initial symptoms typically include bloody diarrhea, fever, vomiting, and weakness.[1][2] Kidney problems and low platelets then occur as the diarrhea progresses.[1] Children are more commonly affected, but most children recover without permanent damage to their health, although some children may have serious and sometimes life-threatening complications.[6] Adults, especially the elderly, may present a more complicated presentation.[2][6] Complications may include neurological problems and heart failure.[1]
| Hemolytic–uremic syndrome | |
|---|---|
| Other names | Haemolytic–uraemic syndrome |
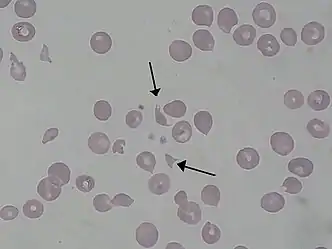 | |
| Schistocytes as seen in a person with hemolytic–uremic syndrome | |
| Specialty | Pediatrics, nephrology |
| Symptoms | Early: Bloody diarrhea, vomiting, fever Later: Low platelets, low red blood cells, kidney failure[1] |
| Complications | Neurological problems, heart failure[1] |
| Types | Shiga toxin–producing E. coli HUS (STEC HUS), S. pneumoniae-associated HUS (SP-HUS), Atypical hemolytic uremic syndrome (aHUS), Cobalamin C HUS[1] |
| Causes | Infection by E coli O157:H7, shigella, salmonella[2] |
| Risk factors | Younger age, female, immunocompromised, or existing renal, urinary, or lower GI disease (because these are the systems involved in the disease)[1] |
| Diagnostic method | Blood tests (to monitor levels of platelets, red blood cells, and white blood cells), stool tests (especially to check for microscopic or macroscopic levels of fresh or old blood), urinalysis (to help monitor kidney function, like blood urea nitrogen, or BUN, levels, pH, and for blood in the urine- hematuria)[3] |
| Differential diagnosis | Thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC), certain problems with an artificial heart valve[4] |
| Treatment | Supportive care, dialysis, steroids, blood transfusions, plasmapheresis[2][1] |
| Prognosis | <25% long-term kidney problems, which for some of these, could include chronic kidney dysfunction or even failure (which could ultimately need dialysis or transplantation to treat);[1] 5% risk of death during the illness in developed countries with treatment |
| Frequency | 1.5 per 100,000 per year[5] |
| Deaths | <5% risk of death[1] |
Most cases occur after infectious diarrhea due to a specific type of E. coli called O157:H7.[2] Other causes include S. pneumoniae, Shigella, Salmonella, and certain medications.[1][2][3] The underlying mechanism typically involves the production of Shiga toxin by the bacteria.[1][2] Atypical hemolytic uremic syndrome (aHUS) is often due to a genetic mutation and presents differently.[1][2] However, both can lead to widespread inflammation and multiple blood clots in small blood vessels, a condition known as thrombotic microangiopathy.[7]
Treatment involves supportive care and may include dialysis, steroids, blood transfusions, or plasmapheresis.[1][2] About 1.5 per 100,000 people are affected per year.[5][1] Less than 5% of those with the condition die.[1] Of the remainder, up to 25% have ongoing kidney problems.[1] HUS was first defined as a syndrome in 1955.[1][8]
Signs and symptoms
After eating contaminated food, the first symptoms of infection can emerge anywhere from 1 to 10 days later, but usually after 3 to 4 days.[9] These early symptoms can include diarrhea (which is often bloody), stomach cramps, mild fever,[10] or vomiting that results in dehydration and reduced urine.[9] HUS typically develops about 5–10 days after the first symptoms, but can take up to 3 weeks to manifest, and occurs at a time when the diarrhea is improving.[10] Related symptoms and signs include lethargy, decreased urine output, blood in the urine, kidney failure, low platelets, (which are needed for blood clotting), and destruction of red blood cells (microangiopathic hemolytic anemia). High blood pressure, jaundice (a yellow tinge in skin and the whites of the eyes), seizures, and bleeding into the skin can also occur.[10] In some cases, there are prominent neurologic changes.[11][12][13]
People with HUS commonly exhibit the symptoms of thrombotic microangiopathy (TMA), which can include abdominal pain,[14] low platelet count,[15] elevated lactate dehydrogenase LDH, (an enzyme released from damaged cells, and which is therefore a marker of cellular damage)[16] decreased haptoglobin (indicative of the breakdown of red blood cells)[16] anemia (low red blood cell count), schistocytes (damaged red blood cells),[15][16] elevated creatinine (a protein waste product generated by muscle metabolism and eliminated renally),[17] proteinuria (indicative of kidney injury),[18] confusion,[14] fatigue,[19] swelling,[20] nausea/vomiting,[21] and diarrhea.[22] Additionally, patients with aHUS typically present with an abrupt onset of systemic signs and symptoms such as acute kidney failure,[15] hypertension (high blood pressure),[19] myocardial infarction (heart attack),[23] stroke,[14] lung complications,[23] pancreatitis (inflammation of the pancreas),[21] liver necrosis (death of liver cells or tissue),[15][19] encephalopathy (brain dysfunction),[19] seizure,[24] and coma.[25] Failure of neurologic, cardiac, renal, and gastrointestinal (GI) organs, as well as death, can occur unpredictably at any time, either very quickly or following prolonged symptomatic or asymptomatic disease progression.[5][7][15][18][26]
Cause
Typical HUS
Shiga-toxin producing E. coli (STEC) HUS occurs after ingestion of a strain of bacteria expressing Shiga toxin such as enterohemorrhagic Escherichia coli (EHEC), of which E. coli O157:H7 is the most common serotype.[27]
Atypical HUS
Atypical HUS (aHUS) represents 5–10% of HUS cases[5] and is largely due to one or several genetic mutations that cause chronic, uncontrolled, and excessive activation of the complement system,[5] which is a group of immune signaling factors that promote inflammation, enhance the ability of antibodies and phagocytic cells to clear microbes and damaged cells from the body, and directly attack the pathogen's cell membrane. This results in platelet activation, endothelial cell damage, and white blood cell activation, leading to systemic TMA, which manifests as decreased platelet count, hemolysis (breakdown of red blood cells), damage to multiple organs, and ultimately death.[7][18][28] Early signs of systemic complement-mediated TMA include thrombocytopenia (platelet count below 150,000 or a decrease from baseline of at least 25%)[16] and evidence of microangiopathic hemolysis, which is characterized by elevated LDH levels, decreased haptoglobin, decreased hemoglobin (the oxygen-containing component of blood), and/or the presence of schistocytes.[7][29][16] Despite the use of supportive care, an estimated 33–40% of patients will die or have end-stage renal disease (ESRD) with the first clinical manifestation of aHUS,[22][23] and 65% of patients will die, require dialysis, or have permanent renal damage within the first year after diagnosis despite plasma exchange or plasma infusion (PE/PI) therapy.[22] Patients who survive the presenting signs and symptoms of aHUS endure a chronic thrombotic and inflammatory state, which puts them at lifelong elevated risk of sudden blood clotting, kidney failure, other severe complications and premature death.[29][20]
Historically, treatment options for aHUS were limited to plasma exchange or plasma infusion (PE/PI) therapy, which carries significant risks[30][31] and has not been proven effective in any controlled trials. People with aHUS and ESRD have also had to undergo lifelong dialysis, which has a 5-year survival rate of 34–38%.[32][33]
Pathogenesis
HUS is caused by ingestion of bacteria that produce Shiga toxins, with Shiga-toxin producing E. coli (STEC) being the most common type.[34] E. coli can produce shigatoxin-1, shigatoxin-2, or both; with shigatoxin-2 producing organisms being more virulent and being much more likely to cause HUS.[34] Once ingested, the bacteria move to the intestines where the produce the Shiga toxins. The bacteria and toxins damage the mucosal lining of the intestines, and thus are able to gain entry into the circulation.[34] Shiga toxin enters the mesenteric microvasculature lining the intestines where it releases inflammatory cytokines including IL-6, IL-8, TNFα, and IL-1β.[34] These inflammatory mediators lead to inflammation and vascular injury with microthrombi that are seen with HUS. It also further damages the intestinal barrier leading to diarrhea (usually bloody) and further entry of Shiga toxin from the intestines to the bloodstream as the intestinal barrier is compromised.[34]
Once Shiga toxin enters the circulation it can travel throughout the body and cause the wide array of end organ damage and the multitude of symptoms seen with HUS. Shiga toxin gains entry to cells by binding to globotriaosylceramide (Gb3) which is a globoside found on cell membranes, it is found throughout the body including the surface of the glomerular endothelium of the kidney.[35] Shiga toxin gains entry to the cell via Gb3 and endocytosis, it then is transported to the Golgi apparatus where furin cleaves the A subunit of the Shiga toxin.[34] It is then transported to the endoplasmic reticulum where it is further cleaved, leaving the A1 subunit of Shiga toxin free. The A1 subunit of Shiga toxin inhibits the 28s subunit of the ribosomal rRNA, this leads to inhibited protein production by the ribosomes.[34] With the cell's protein synthesis inhibited by Shiga toxin, the cell is destroyed.[34] This leads to vascular injury (including in the kidneys where Gb3 is concentrated). The vascular injury facilitates the formation of vascular microthrombi which are characteristic of TTP.[34] The TTP leads to platelet trapping (and thrombocytopenia), red blood cell destruction (and anemia), and end organ damage that is characteristically seen with HUS and TTP.[34]
HUS is one of the thrombotic microangiopathies, a category of disorders that includes STEC-HUS, aHUS, and thrombotic thrombocytopenic purpura (TTP). The release of cytokines and chemokines (IL-6, IL-8, TNF-α, IL-1β) that are commonly released by Shiga toxin are implicated in platelet activation and TTP.[36] The presence of schistocytes is a key finding that helps to diagnose HUS.
Shiga-toxin directly activates the alternative complement pathway and also interferes with complement regulation by binding to complement factor H, an inhibitor of the complement cascade. Shiga-toxin causes complement-mediated platelet, leukocyte, and endothelial cell activation, resulting in systemic hemolysis, inflammation and thrombosis.[37][38][39] Severe clinical complications of TMA have been reported in patients from 2 weeks to more than 44 days after presentation with STEC-HUS, with improvements in clinical condition extending beyond this time frame, suggesting that complement activation persists beyond the acute clinical presentation and for at least 4 months.[40]
The consumption of platelets as they adhere to the thrombi lodged in the small vessels typically leads to mild or moderate thrombocytopenia with a platelet count of less than 60,000 per microliter.[41] As in the related condition TTP, reduced blood flow through the narrowed blood vessels of the microvasculature leads to reduced blood flow to vital organs, and ischemia may develop.[11] The kidneys and the central nervous system (brain and spinal cord) are the parts of the body most critically dependent on high blood flow, and are thus the most likely organs to be affected. However, in comparison to TTP, the kidneys tend to be more severely affected in HUS, and the central nervous system is less commonly affected.[42]
In contrast with typical disseminated intravascular coagulation seen with other causes of sepsis and occasionally with advanced cancer, coagulation factors are not consumed in HUS (or TTP) and the coagulation screen, fibrinogen level, and assays for fibrin degradation products such as "D-Dimers", are generally normal despite the low platelet count (thrombocytopenia).[42]
HUS occurs after 3–7% of all sporadic E. coli O157:H7 infections and up to approximately 20% or more of epidemic infections.[43] Children and adolescents are commonly affected.[44] One reason could be that children have more Gb3 receptors than adults which may be why children are more susceptible to HUS. Cattle, swine, deer, and other mammals do not have GB3 receptors, but can be asymptomatic carriers of Shiga toxin-producing bacteria. Some humans can also be asymptomatic carriers. Once the bacteria colonizes, diarrhea followed by bloody diarrhea, hemorrhagic colitis, typically follows. Other serotypes of STEC also cause disease, inlduding HUS, as occurred with E. coli O104:H4, which triggered a 2011 epidemic of STEC-HUS in Germany.[45]
Grossly, the kidneys may show patchy or diffuse renal cortical necrosis. Histologically, the glomeruli show thickened and sometimes split capillary walls due largely to endothelial swelling. Large deposits of fibrin-related materials in the capillary lumens, subendothelially, and in the mesangium are also found along with mesangiolysis. Interlobular and afferent arterioles show fibrinoid necrosis and intimal hyperplasia and are often occluded by thrombi.[12]
STEC-HUS most often affects infants and young children, but also occurs in adults. The most common form of transmission is ingestion of undercooked meat, unpasteurized fruits and juices, contaminated produce, contact with unchlorinated water, and person-to-person transmission in daycare or long-term care facilities.[25]
Unlike typical HUS, aHUS does not follow STEC infection and is thought to result from one or several genetic mutations that cause chronic, uncontrolled, and excessive activation of complement.[5] This leads to platelet activation, endothelial cell damage, and white blood cell activation, leading to systemic TMA, which manifests as decreased platelet count, hemolysis, damage to multiple organs, and ultimately, death.[7][18][28] Early signs of systemic complement-mediated TMA include thrombocytopenia (platelet count below 150,000 or a decrease from baseline of at least 25%)[16] and evidence of microangiopathic hemolysis, which is characterized by elevated LDH levels, decreased haptoglobin, decreased hemoglobin, and/or the presence of schistocytes.[7][29][16]
Diagnosis
The similarities between HUS, aHUS, and TTP make differential diagnosis essential.[7][29] All three of these systemic TMA-causing diseases are characterized by thrombocytopenia[16] and microangiopathic hemolysis,[5][16] plus one or more of the following: neurological symptoms (e.g., confusion,[5][24] cerebral convulsions,[24] seizures[21]); renal impairment[16] (e.g., elevated creatinine,[17] decreased estimated glomerular filtration rate [eGFR],[17] abnormal urinalysis[46]); and gastrointestinal (GI) symptoms (e.g., diarrhea,[19][22] nausea/vomiting,[21] abdominal pain,[21] gastroenteritis[16][19]).The presence of diarrhea does not exclude aHUS as the cause of TMA, as 28% of patients with aHUS present with diarrhea and/or gastroenteritis.[18][19] First diagnosis of aHUS is often made in the context of an initial, complement-triggering infection, and Shiga-toxin has also been implicated as a trigger that identifies patients with aHUS.[40] Additionally, in one study, mutations of genes encoding several complement regulatory proteins were detected in 8 of 36 (22%) patients diagnosed with STEC-HUS.[47] However, the absence of an identified complement regulatory gene mutation does not preclude aHUS as the cause of the TMA, as approximately 50% of patients with aHUS lack an identifiable mutation in complement regulatory genes.[19]
Diagnostic work-up supports the differential diagnosis of TMA-causing diseases. A positive Shiga-toxin/EHEC test confirms a cause for STEC-HUS,[25][27] and severe ADAMTS13 deficiency (i.e., ≤5% of normal ADAMTS13 levels) confirms a diagnosis of TTP.[48]
Prevention
The effect of antibiotics in shiga toxin producing E. coli is unclear.[1] While some early studies raised concerns more recent studies show either no effect or a benefit.[1]
Treatment
Treatment involves supportive care and may include dialysis, steroids, blood transfusions, and plasmapheresis.[1][2] Early IV fluid hydration is associated with better outcomes including shorter hospital stays and reducing the risk of dialysis.[34]
Empiric antibiotics are not indicated in those who are immunocompetent, and may worsen the HUS.[34] Antidiarrheals and narcotic medications to slow the gut are not recommended as they are associated with worsening symptoms, increased risk of HUS in those with STEC infection, and adverse neurologic reactions.[34] Platelet transfusions should not be used as the may drive the process of microangiopathy leading to worsening TTP.[34]
While eculizumab is being used to treat atypical hemolytic uremic syndrome, no evidence as of 2018 supports its use in the main forms of HUS.[1] Scientists are trying to understand how useful it would be to immunize humans or cattle.[49]
Prognosis
Acute renal failure occurs in 55–70% of people with STEC-HUS, although up to 70–85% recover renal function.[50] With aggressive treatment, more than 90% of patients survive the acute phase of HUS, and only about 9% may develop ESRD. Roughly one-third of persons with HUS have abnormal kidney function many years later, and a few require long-term dialysis. Another 8% of persons with HUS have other lifelong complications, such as high blood pressure, seizures, blindness, paralysis, and the effects of having part of their colon removed. STEC-HUS is associated with a 3% mortality rate among young children and a 20% mortality rate in middle age or older adults.[34] 15-20% of children infected with STEC develop HUS, with the highest risk being in children younger than 5 years old.[34]
Patients with aHUS generally have poor outcomes, with up to 50% progressing to end-stage renal disease (ESRD) or irreversible brain damage; as many as 25% die during the acute phase.[50]
History
HUS is now considered as a part of the broader group of Thrombotic microangiopathies (TMA). Thrombotic thrombocytopenic purpura (TTP), a TMA, was first described by the Hungarian born, American pathologist and physician Eli Moschcowitz (1879–1964). In 1924,[51] Moschcowitz first described TTP as a distinct clinicopathologic condition that can mimic the clinical characteristics of Hemolytic–uremic syndrome (HUS). That was in a 16-year-old girl who died 2 weeks after the abrupt onset and progression of petechial bleeding, pallor, fever, paralysis, hematuria and coma; and called “Moschcowitz disease”.[52][53] Moreover, Moschcowitz was among the first to work in psychosomatic medicine, and he presented a paper in 1935 on the psychological origins of physical disease. HUS was first described by Conrad Gasser in 1955, and the systemic character of HUS was subsequently defined.[54] Bernard Kaplan identified several distinct entities that can manifest as HUS and emphasized that HUS was a syndrome with a common pathologic outcome. Kaplan is a Canadian professor and director of Pediatric Nephrology. He has an international reputation for his studies, over the past 34 years, on the hemolytic uremic syndromes.[55] The discovery that endothelial cell injury underlies this broad spectrum of TMA disorders has come into focus during the last two decades. In the 1980s, Mohamed Karmali (1945-2016) was the first to make the association between Stx, diarrheal E. coli infection and the idiopathic hemolytic uremic syndrome of infancy and childhood. Karmali’s work showed that the hemolytic uremic syndrome the children in Canada was caused by this particular bacteria. Karmali also developed the system of classifying strains of E.coli and determining which cause disease in humans. He defined the presence of microvascular injury in diarrhea-associated HUS and the critical role of a verotoxin produced by specific strains of Escherichia coli.[56] This verotoxin was subsequently found to be a member of a family of toxins first identified with Shigella and known as Shiga toxin (Stx).[57] This relationship and the eventual link of TTP to abnormally high levels of ultra-large Von Willebrand factor (vWF) multimers caused by congenital or acquired reductions in ADAMTS13 activity was established at approximately the same time. In 1924, a Finnish physician Erik Adolf von Willebrand (1870-1949) was consulted about a young girl with a bleeding disorder. Von Willebrand[58] described this disorder in 1926, distinguishing it from hemophilia. The disorder was named after him, becoming known as von Willebrand disease. The cause of the disease was later discovered to be a deficiency of a protein, now known as von Willebrand factor, that enables hemostasis. Paul Warwicker is an English nephrologist, whilst in Newcastle in the mid 1990s his research in molecular genetics with Professors Tim and Judith Goodship led to the genetic mapping of the familial form of atypical HUS and the descriptions of the first HUS-related mutations and polymorphisms in the factor H gene in both familial and sporadic HUS. He was awarded an MD in molecular genetics in 2000, and elected fellow of the Royal College of Physicians in the same year.[59] Paul Warwicker confirmed the association of atypical HUS (aHUS) to defects in a region on chromosome 1 that contains the genes for several complement regulatory proteins.[60] Later, mutations in complement factor H, complement factor I, membrane cofactor protein, factor B, C3, and thrombomodulin have now been found to cause many of the familial cases of aHUS. These discoveries have allowed a more comprehensive understanding of the pathogenesis, evaluation, and treatment of the entire spectrum of TMA disorders and provide a more rational and effective approach to the care of these children with complicated disease. Prior to the use of monoclonal antibodies patients with aHUS had an extremely poor prognosis. Eculizumab (Soliris®, Alexion Pharmaceuticals, Inc., Boston, MA, USA) is a humanized monoclonal complement inhibitor that is the first and only approved treatment for patients with aHUS by FDA in September 2011. Eculizumab binds with high affinity to C5, inhibiting C5 cleavage to C5a and C5b and preventing the generation of the terminal complement complex C5b-9, thus inhibiting complement-mediated TMA. Eculizumab was proven to be effective in patients with aHUS in which it resolved and prevented complement-mediated TMA, improving renal function and hematologic outcomes.[61] Alexion head of R&D 'John Orloff, M.D. “The results met the high bar of complete TMA response, defined by hematologic normalization and improved kidney function,” said Alexion R&D head John Orloff, M.D., who reckons the drug can become the “new standard of care for patients with aHUS.” “We are preparing regulatory submissions for Ultomiris in aHUS in the U.S., European Union and Japan as quickly as possible,” he added.[62]
Epidemiology
The country with the highest incidence of HUS is Argentina[63][64][65][66] and it performs a key role in the research of this condition.
In the United States, the overall incidence of HUS is estimated at 2.1 cases per 100,000 persons/year, with a peak incidence between six months and four years of age.[67]
HUS and the E. coli infections that cause it have been the source of much negative publicity for the FDA, meat industries, and fast-food restaurants since the 1990s, especially in the contaminations linked to Jack in the Box restaurants. In 2006, an epidemic of harmful E. coli emerged in the United States due to contaminated spinach. In June 2009, Nestlé Toll House cookie dough was linked to an outbreak of E. coli O157:H7 in the United States, which sickened 70 people in 30 states.[67]
In May 2011 an epidemic of bloody diarrhea caused by E. coli O104:H4-contaminated fenugreek seeds hit Germany. Tracing the epidemic revealed more than 3,800 cases, with HUS developing in more than 800 of the cases, including 36 fatal cases. Nearly 90% of the HUS cases were in adults.[68][69]
References
- Cody, EM; Dixon, BP (February 2019). "Hemolytic Uremic Syndrome". Pediatric Clinics of North America. 66 (1): 235–246. doi:10.1016/j.pcl.2018.09.011. PMID 30454746. S2CID 53875876.
- "Hemolytic uremic syndrome". Genetic and Rare Diseases Information Center (GARD). Retrieved 21 November 2018.
- Salvadori, M; Bertoni, E (6 August 2013). "Update on hemolytic uremic syndrome: Diagnostic and therapeutic recommendations". World Journal of Nephrology. 2 (3): 56–76. doi:10.5527/wjn.v2.i3.56. PMC 3832913. PMID 24255888.
- Ferri, Fred F. (2010). Ferri's Differential Diagnosis E-Book: A Practical Guide to the Differential Diagnosis of Symptoms, Signs, and Clinical Disorders. Elsevier Health Sciences. p. 219. ISBN 978-0323081634.
- Noris, M; Remuzzi, G (2009). "Atypical hemolytic–uremic syndrome". N Engl J Med. 361 (17): 1676–1687. doi:10.1056/NEJMra0902814. PMID 19846853.
- Chu, P; Hemphill, RR (2004). "222: Acquired hemolytic anemia". Emergency Medicine: A Comprehensive Study Guide (6th ed.). New York, NY: McGraw-Hill. ISBN 978-0-07-138875-7.
- Benz, K; Amann, K (2010). "Thrombotic microangiopathy: new insights". Current Opinion in Nephrology and Hypertension. 19 (3): 242–247. doi:10.1097/MNH.0b013e3283378f25. PMID 20186056. S2CID 25429151.
- Gasser C, Gautier E, Steck A, Siebenmann RE, Oechslin R (September 1955). "Hemolytic–uremic syndrome: bilateral necrosis of the renal cortex in acute acquired hemolytic anemia". Schweiz Med Wochenschr (in German). 85 (38–39): 905–9. PMID 13274004.
- "E.coli (Escherichia coli): Symptoms". Centers for Disease Control and Prevention. U.S. Department of Health & Human Services. 2017-11-30. Retrieved 22 November 2018.
- "Hemolytic uremic syndrome (HUS)". Center for Acute Disease Epidemiology. Iowa Department of Public Health. Retrieved 21 November 2018.
- Boyer, O; Niaudet, P (August 2011). "Hemolytic Uremic Syndrome: New Developments in Pathogenesis and Treatments". Int J Nephrol. 2011: 908407. doi:10.4061/2011/908407. PMC 3159990. PMID 21876803.
- Kumar, V; Cotran, RS; Robbins, SL, eds. (2002). Robbins Basic Pathology. Philadelphia, PA: Saunders. ISBN 978-0721692746.
- Nathanson, S.; Kwon, T.; Elmaleh, M.; et al. (2010). "Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome". Clin J Am Soc Nephrol. 5 (7): 1218–1228. doi:10.2215/CJN.08921209. PMC 2893076. PMID 20498239.
- Ohanian, M; Cable, C; Halka, K (2011). "Eculizumab safely reverses neurologic impairment and eliminates the need for dialysis in severe atypical hemolytic uremic syndrome". Clin Pharmacol. 3: 5–12. doi:10.2147/CPAA.S17904. PMC 3262387. PMID 22287852.
- Loirat, C; Noris, M; Fremaux-Bacchi, V (2008). "Complement and the atypical hemolytic uremia syndrome in children". Pediatr Nephrol. 23 (11): 1957–1972. doi:10.1007/s00467-008-0872-4. PMC 6904381. PMID 18594873.
- Caprioli, J.; Noris, M.; Brioschi, S.; et al. (2006). "Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome". Blood. 108 (4): 1267–1279. doi:10.1182/blood-2005-10-007252. PMC 1895874. PMID 16621965.
- Ariceta, Gema; Besbas, Nesrin; Johnson, Sally; et al. (2009). "Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome". Pediatr Nephrol. 24 (4): 687–696. doi:10.1007/s00467-008-0964-1. PMID 18800230.
- Sellier-Leclers, A.-L.; Fremeaux-Bacchi, V.; Dragon-Durey, M.-A.; et al. (2007). "Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome". J Am Soc Nephrol. 18 (8): 2392–2400. doi:10.1681/ASN.2006080811. PMID 17599974.
- Noris, M.; Caprioli, J.; Bresin, E.; et al. (2010). "Relative role of genetic complement abnormalities in infrequent and familial aHUS and their impact on clinical phemotype". Clin J Am Soc Nephrol. 5 (10): 1844–1859. doi:10.2215/CJN.02210310. PMC 2974386. PMID 20595690.
- Ståhl, A.-L.; Vazir-Sani, F.; Heinen, S.; et al. (2008). "Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation". Blood. 111 (11): 5307–5317. doi:10.1182/blood-2007-08-106153. PMID 18268093.
- Dragon-Durey, M.-A.; Sethi, S. K.; Bagga, A.; et al. (2010). "Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome". J Am Soc Nephrol. 21 (12): 2180–2187. doi:10.1681/ASN.2010030315. PMC 3014031. PMID 21051740.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Zuber, J; Le Quintrec, M; Sberro-Scussan, R; Loirat, C; Fremaux-Bacchi, V; Legendre, C (2011). "New insights into postrenal transplant hemolytic uremic syndrome". Nature Reviews Nephrology. 7 (1): 23–35. doi:10.1038/nrneph.2010.155. PMID 21102542. S2CID 2054556.
- Sallee, M; et al. (2010). "Myocardial infarction is a complication of factor H-associated atypical HUS". Nephrol Dial Transplant. 25 (6): 2028–2032. doi:10.1093/ndt/gfq160. PMID 20305136.
- Neuhaus, TJ; Calonder, S; Leumann, EP (1997). "Heterogeneity of atypical haemolytic uraemis syndromes". Arch Dis Child. 76 (6): 518–521. doi:10.1136/adc.76.6.518. PMC 1717216. PMID 9245850.
- Noris, M; Remuzzi, G (2005). "Hemolytic uremic syndrome". J Am Soc Nephrol. 16 (4): 1035–1050. doi:10.1681/ASN.2004100861. PMID 15728781.
- Mache, CJ; et al. (2009). "Complement inhibitor eculizumab in atypical hemolytic uremic syndrome". Clin J Am Soc Nephrol. 4 (8): 1312–1316. doi:10.2215/CJN.01090209. PMC 2723971. PMID 19556379.
- Palermo, MS; Exeni, RA; Fernandez, GC (2009). "Hemolytic Uremic Syndrome: pathogenesis and update of interventions". Expert Rev Anti Infect Ther. 7 (6): 697–707. doi:10.1586/eri.09.49. PMID 19681698. S2CID 30420020.
- Tsai, H-M (2010). "Pathophysiology of thrombotic thrombocytopenic purpura". Int J Hematol. 91 (1): 1–19. doi:10.1007/s12185-009-0476-1. PMC 3159000. PMID 20058209.
- Zipfel, PF; Heinen, S; Skerka, C (2010). "Thrombotic microangiopathies: new insights and new challenges". Current Opinion in Nephrology and Hypertension. 19 (4): 372–378. doi:10.1097/MNH.0b013e32833aff4a. PMID 20539230. S2CID 28419.
- George, JN (2010). "How I treat my patients with thrombotic thrombocytopenic purpura". Blood. 116 (20): 4060–4069. doi:10.1182/blood-2010-07-271445. PMID 20686117. S2CID 26844964.
- Michon, B; et al. (2007). "Complications of apheresis in children". Transfusion. 47 (10): 1837–1842. doi:10.1111/j.1537-2995.2007.01405.x. PMID 17880609. S2CID 23613105.
- Collins, Allan J.; et al. (2010). "Excerpts from the US Renal Data System 2009 Annual Data Report". Am J Kidney Dis. 55 (1 Suppl 1): S1–S7. doi:10.1053/j.ajkd.2009.10.009. PMC 2829836. PMID 20082919.
- European Renal Association- European Dialysis and Transplant Association Registry (2011). ERA-EDTA Registry Annual Report 2009. Amsterdam, the Netherlands: Academic Medical Center Department of Medical Informatics.
- Freedman, Stephen B.; van de Kar, Nicole C.A.J.; Tarr, Phillip I. (12 October 2023). "Shiga Toxin–Producing Escherichia coli and the Hemolytic–Uremic Syndrome". New England Journal of Medicine. 389 (15): 1402–1414. doi:10.1056/NEJMra2108739.
- Psotka, MA; et al. (2009). "Shiga toxin 2 targets the murine renal collecting duct epithelium". Infect Immun. 77 (3): 959–969. doi:10.1128/IAI.00679-08. PMC 2643625. PMID 19124603.
- Guessous, F; et al. (2005). "Shiga toxin 2 and lipopolysaccharide induce human microvascular endothelial cells to release chemokines and factor that stimulate platelet function". Infect Immun. 73 (12): 8306–8316. doi:10.1128/IAI.73.12.8306-8316.2005. PMC 1307066. PMID 16299328.
- Orth D, Würzner R (September 2010). "Complement in typical hemolytic uremic syndrome". Seminars in Thrombosis and Hemostasis. 36 (6): 620–4. doi:10.1055/s-0030-1262883. PMID 20865638. S2CID 260321080.
- Stahl, AL; Startz, L; Karpman, D (2011). "Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome". Blood. 117 (20): 5503–5513. doi:10.1182/blood-2010-09-309161. PMID 21447825.
- Thurman, J.M.; et al. (2009). "Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome". Clin J Am Soc Nephrol. 4 (12): 1920–1924. doi:10.2215/CJN.02730409. PMC 2798880. PMID 19820137.
- Mache, C; et al. (June 2010). "Eculizumab in diarrhea-associated hemolytic uremic syndrome". Presented at the 2nd International Conference on HUS-MPGN-PNH.
- Tan, AJ (2017-12-27). "Hemolytic uremic syndrome in emergency medicine". Medscape EMedicine.
- Kanso, AA; Abou Hassan, NM; Badr, KF (2008). "Micro and macrovascular disease of the kidney". In: Brenner BM, ed. The Kidney, 8th Edition: chapter 32.
- Mead, PS; Griffin, PM (1998). "Escherichia coli O157:H7". Lancet. 352 (9135): 1207–1212. doi:10.1016/S0140-6736(98)01267-7. PMID 9777854. S2CID 38100845.
- Ruggenenti, P; Noris, M; Remuzzi, G (2001). "Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura". Kidney Int. 60 (3): 831–846. doi:10.1046/j.1523-1755.2001.060003831.x. PMID 11532079.
- Robert Koch Institute. "Technical Report: EHEC/HUS O104:H4 Outbreak, Germany, May/June 2011" (PDF). Berlin, Germany: Robert Koch Institute. Archived from the original (PDF) on 2016-04-08.
- Al-Akash, AI; Almond, PS; Savell, VH Jr; Gharaybeth, SI; Hogue, C (2011). "Eculizumab includes long-term remission in recurrent post-transplant HUS associated with C3 gene mutation". Pediatr Nephrol. 26 (4): 613–619. doi:10.1007/s00467-010-1708-6. PMID 21125405. S2CID 22334044.
- Gangnadoux, MF; Habib, R; Gubler, MC; Bacri, JL; Broyer, M (1996). "Long-term (15-25 years) outcome of childhood hemolytic–uremic syndrome". Clin Nephrol. 46 (1): 39–41. PMID 8832149.
- Zhen, XL; et al. (2010). "Multiple domains of ADAMTS13 are targeted by autoantibodies against ADAMTS13 in patients with acquired idiopathic thrombotic thrombocytopenic purpura". Haematologica. 95 (9): 1555–1562. doi:10.3324/haematol.2009.019299. PMC 2930958. PMID 20378566.
- O'Ryan, M; Vidal, R; Del Canto, F; Salazar, J C; Montero, D (2015). "Vaccines for viral and bacterial pathogens causing acute gastroenteritis: Part II: Vaccines for Shigella, Salmonella, enterotoxigenic E. coli (ETEC) enterohemorragic E. coli (EHEC) and Campylobacter jejuni". Human Vaccines & Immunotherapeutics. 11 (3): 601–619. doi:10.1080/21645515.2015.1011578. PMC 4514228. PMID 25715096.
- Parmar, MS (2010). "Hemolytic–uremic syndrome". Medscape EMedicine.
- Sukumar S, Lämmle B, Cataland SR (January 2021). "Thrombotic Thrombocytopenic Purpura: Pathophysiology, Diagnosis, and Management". Journal of Clinical Medicine. 10 (3): 536. doi:10.3390/jcm10030536. ISSN 2077-0383. PMC 7867179. PMID 33540569.
- Moschcowitz, Eli (1925). "An Acute Febrile Pleiochromic Anemia with Hyaline Thrombosis of the Terminal Arterioles and Capillaries: An Undescribed Disease". Archives of Internal Medicine. 36 (1): 89–93. doi:10.1001/archinte.1925.00120130092009. ISSN 0730-188X:
- —— (1952). "An Acute Febrile Pleiochromic Anemia with Hyaline Thrombosis of the Terminal Arterioles and Capillaries: An Undescribed Disease". The American Journal of Medicine. 13 (5): 567–569. doi:10.1016/0002-9343(52)90022-3. ISSN 0002-9343. PMID 12996533.
- —— (1978). "An Acute Febrile Pleiochromic Anemia with Hyaline Thrombosis of the Terminal Arterioles and Capillaries: An Undescribed Disease". Thrombosis and Haemostasis. 40 (4): 004–008. doi:10.1055/s-0039-1681113. ISSN 0340-6245. PMID 725850. S2CID 29544299.
- —— (2003). "An Acute Febrile Pleiochromic Anemia with Hyaline Thrombosis of the Terminal Arterioles and Capillaries: An Undescribed Disease". The Mount Sinai Journal of Medicine. 70 (5): 352–355. PMID 14631522.
- Lichtman, Marshall A.; et al., eds. (2000). "Commentary on and reprint of Moschcowitz E, An acute febrile pleiochromic anemia with hyaline thrombosis of the terminal arterioles and capillaries: An undescribed disease, in Archives of Internal Medicine (1925) 36:89–93". Hematology. Academic Press. pp. 119–125. doi:10.1016/B978-012448510-5.50105-9. ISBN 978-0-12-448510-5.
- Gianantonio, Carlos A. (1984), "Hemolytic Uremic Syndrome", Acute Renal Failure, Boston, MA: Springer US, pp. 327–339, doi:10.1007/978-1-4613-2841-4_16, ISBN 978-1-4612-9794-9, S2CID 46876846, retrieved 2023-08-21
- Kaplan, Bernard S.; Drummond, Keith N. (1978-04-27). "The Hemolytic-Uremic Syndrome Is a Syndrome". New England Journal of Medicine. 298 (17): 964–966. doi:10.1056/nejm197804272981710. ISSN 0028-4793. PMID 642978.
- OVC Communications. "Dr. Mohamed Karmali – Solving the Mystery of the Haemolytic-Uremic Syndrome - June 10". Tumblr. Retrieved 2023-08-21.
- Karmali, M. A.; Petric, M.; Lim, C.; Fleming, P. C.; Arbus, G. S.; Lior, H. (1985-05-01). "The Association Between Idiopathic Hemolytic Uremic Syndrome and Infection by Verotoxin-Producing Escherichia coli". Journal of Infectious Diseases. 151 (5): 775–782. doi:10.1093/infdis/151.5.775. ISSN 0022-1899. PMID 3886804.
- "Erik Adolf von Willebrand", Wikipedia, 2023-01-12, retrieved 2023-08-21
- "Dr Paul Warwicker : General (internal) medicine , Renal medicine". www.finder.bupa.co.uk. Retrieved 2023-08-22.
- Warwicker, Paul; Goodship, Timothy H.J.; Donne, Rosemary L.; Pirson, Yves; Nicholls, Anthony; Ward, Roy M.; Turnpenny, Peter; Goodship, Judith A. (April 1998). "Genetic studies into inherited and sporadic hemolytic uremic syndrome". Kidney International. 53 (4): 836–844. doi:10.1111/j.1523-1755.1998.00824.x. ISSN 0085-2538. PMID 9551389.
- "2011 Notifications". FDA. 3 November 2018.
- Taylor, Phil (28 January 2019). "Alexion's Soliris follow-up clears trial in aHUS, its second rare disease". fiercebiotech.com.
- Rivero, MA; Passucci, JA; Rodriguez, EM; Signorini, ML; Tarabla, HD; Parma, AE (2011). "Factors associated with sporadic verotoxigenic Escherichia coli infection in children with diarrhea from the Central Eastern Area of Argentina". Foodborne Pathogens and Disease. 8 (8): 901–6. doi:10.1089/fpd.2010.0800. PMID 21492023.
- Rivas, M; Caletti, MG; Chinen, I; Refi, SM; Roldán, CD; Chillemi, G; Fiorilli, G; Bertolotti, A; Aguerre, L; Sosa Estani, S (2003). "Home-prepared hamburger and sporadic hemolytic uremic syndrome, Argentina". Emerging Infectious Diseases. 9 (9): 1184–6. doi:10.3201/eid0909.020563. PMC 3016759. PMID 14531383.
- Rivero, MA; Padola, NL; Etcheverría, AI; Parma, AE (2004). "Enterohemorrhagic Escherichia coli and hemolytic–uremic syndrome in Argentina". Medicina. 64 (4): 352–6. PMID 15338982.
- "What is HUS?" (PDF).
- Corrigan JJ, Boineau FG (November 2001). "Hemolytic–uremic syndrome". Pediatr Rev. 22 (11): 365–9. doi:10.1542/pir.22-11-365. PMID 11691946.
- Buchholz, U; et al. (2011). "German outbreak of Escherichia coli O104:H4 associated with sprouts". N Engl J Med. 365 (19): 1763–1770. doi:10.1056/NEJMoa1106482. PMID 22029753.
- Frank, C; et al. (2011). "Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany". N Engl J Med. 365 (19): 1711–1780. doi:10.1056/NEJMoa1106483. PMID 21696328. S2CID 205093464.