Variants of PCR
The versatility of polymerase chain reaction (PCR) has led to modifications of the basic protocol being used in a large number of variant techniques designed for various purposes. This article summarizes many of the most common variations currently or formerly used in molecular biology laboratories; familiarity with the fundamental premise by which PCR works and corresponding terms and concepts is necessary for understanding these variant techniques.
Basic modifications
Often only a small modification needs to be made to the standard PCR protocol to achieve a desired goal:
Multiplex-PCR uses several pairs of primers annealing to different target sequences. This permits the simultaneous analysis of multiple targets in a single sample. For example, in testing for genetic mutations, six or more amplifications might be combined. In the standard protocol for DNA fingerprinting, the targets assayed are often amplified in groups of 3 or 4. Multiplex Ligation-dependent Probe Amplification (MLPA) permits multiple targets to be amplified using only a single pair of primers, avoiding the resolution limitations of multiplex PCR. Multiplex PCR has also been used for analysis of microsatellites and SNPs.[1]

Variable Number of Tandem Repeats (VNTR) PCR targets repetitive areas of the genome that exhibit length variation. Analysis of the genotypes in the samples usually involves sizing of the amplification products by gel electrophoresis. Analysis of smaller VNTR segments known as short tandem repeats (or STRs) is the basis for DNA fingerprinting databases such as CODIS.
Asymmetric PCR preferentially amplifies one strand of a double-stranded DNA target. It is used in some sequencing methods and hybridization probing to generate one DNA strand as product. Thermocycling is carried out exactly as in conventional PCR, but with a limiting amount or leaving out one of the primers. When the limiting primer becomes depleted, replication increases arithmetically rather than exponentially through extension of the excess primer.[2] A modification of this process, named Linear-After-The-Exponential-PCR (or LATE-PCR), uses a limiting primer with a higher melting temperature (Tm) than the excess primer in order to maintain reaction efficiency as the limiting primer concentration decreases mid-reaction.[3] See also overlap-extension PCR.
Some modifications are needed to perform long PCR. The original Klenow-based PCR process did not generate products that were larger than about 400 bp. Taq polymerase can however amplify targets of up to several thousand bp long.[4] Since then, modified protocols with Taq enzyme have allowed targets of over 50 kb to be amplified.[5]
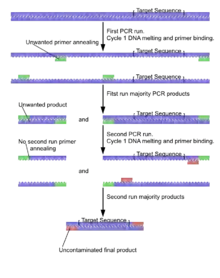
Nested PCR is used to increase the specificity of DNA amplification. Two sets of primers are used in two successive reactions. In the first PCR, one pair of primers is used to generate DNA products, which may contain products amplified from non-target areas. The products from the first PCR are then used as template in a second PCR, using one ('hemi-nesting') or two different primers whose binding sites are located (nested) within the first set, thus increasing specificity. Nested PCR is often more successful in specifically amplifying long DNA products than conventional PCR, but it requires more detailed knowledge of the sequence of the target.
Quantitative PCR (qPCR) is used to measure the specific amount of target DNA (or RNA) in a sample. By measuring amplification only within the phase of true exponential increase, the amount of measured product more accurately reflects the initial amount of target. Special thermal cyclers are used that monitor the amount of product during the amplification.
Quantitative Real-Time PCR (QRT-PCR), sometimes simply called Real-Time PCR (RT-PCR), refers to a collection of methods that use fluorescent dyes, such as Sybr Green, or fluorophore-containing DNA probes, such as TaqMan, to measure the amount of amplified product in real time as the amplification progresses.
Hot-start PCR is a technique performed manually by heating the reaction components to the DNA melting temperature (e.g. 95 °C) before adding the polymerase. In this way, non-specific amplification at lower temperatures is prevented.[6] Alternatively, specialized reagents inhibit the polymerase's activity at ambient temperature, either by the binding of an antibody, or by the presence of covalently bound inhibitors that only dissociate after a high-temperature activation step. 'Hot-start/cold-finish PCR' is achieved with new hybrid polymerases that are inactive at ambient temperature and are only activated at elevated temperatures.
In touchdown PCR, the annealing temperature is gradually decreased in later cycles. The annealing temperature in the early cycles is usually 3–5 °C above the standard Tm of the primers used, while in the later cycles it is a similar amount below the Tm. The initial higher annealing temperature leads to greater specificity for primer binding, while the lower temperatures permit more efficient amplification at the end of the reaction.[7]
Assembly PCR (also known as Polymerase Cycling Assembly or PCA) is the synthesis of long DNA structures by performing PCR on a pool of long oligonucleotides with short overlapping segments, to assemble two or more pieces of DNA into one piece. It involves an initial PCR with primers that have an overlap and a second PCR using the products as the template that generates the final full-length product. This technique may substitute for ligation-based assembly.[8]
In colony PCR, bacterial colonies are screened directly by PCR, for example, the screen for correct DNA vector constructs. Colonies are sampled with a sterile pipette tip and a small quantity of cells transferred into a PCR mix. To release the DNA from the cells, the PCR is either started with an extended time at 95 °C (when standard polymerase is used), or with a shortened denaturation step at 100 °C and special chimeric DNA polymerase.[9]
The digital polymerase chain reaction simultaneously amplifies thousands of samples, each in a separate droplet within an emulsion.
Suicide PCR is typically used in paleogenetics or other studies where avoiding false positives and ensuring the specificity of the amplified fragment is the highest priority. It was originally described in a study to verify the presence of the microbe Yersinia pestis in dental samples obtained from 14th-century graves of people supposedly killed by plague during the medieval Black Death epidemic.[10] The method prescribes the use of any primer combination only once in a PCR (hence the term "suicide"), which should never have been used in any positive-control PCR reaction, and the primers should always target a genomic region never amplified before in the lab using this or any other set of primers. This ensures that no contaminating DNA from previous PCR reactions is present in the lab, which could otherwise generate false positives.
COLD-PCR (co-amplification at lower denaturation temperature-PCR) is a modified protocol that enriches variant alleles from a mixture of wild-type and mutation-containing DNA samples.
Pretreatments and extensions
The basic PCR process can sometimes precede or follow another technique.
RT-PCR (or Reverse Transcription PCR) is used to reverse-transcribe and amplify RNA to cDNA. PCR is preceded by a reaction using reverse transcriptase, an enzyme that converts RNA into cDNA. The two reactions may be combined in a tube, with the initial heating step of PCR being used to inactivate the transcriptase.[4] The Tth polymerase (described below) has RT activity, and can carry out the entire reaction. RT-PCR is widely used in expression profiling, which detects the expression of a gene. It can also be used to obtain sequence of an RNA transcript, which may aid the determination of the transcription start and termination sites (by RACE-PCR) and facilitate mapping of the location of exons and introns in a gene sequence.
Two-tailed PCR uses a single primer that binds to a microRNA target with both 3' and 5' ends, known as hemiprobes.[11] Both ends must be complementary for binding to occur. The 3'-end is then extended by reverse transcriptase forming a long cDNA. The cDNA is then amplified using two target specific PCR primers. The combination of two hemiprobes, both targeting the short microRNA target, makes the Two-tailed assay exceedingly sensitive and specific.
Ligation-mediated PCR uses small DNA oligonucleotide 'linkers' (or adaptors) that are first ligated to fragments of the target DNA. PCR primers that anneal to the linker sequences are then used to amplify the target fragments. This method is deployed for DNA sequencing, genome walking, and DNA footprinting.[12] A related technique is amplified fragment length polymorphism, which generates diagnostic fragments of a genome.
Methylation-specific PCR (MSP) is used to identify patterns of DNA methylation at cytosine-guanine (CpG) islands in genomic DNA.[13] Target DNA is first treated with sodium bisulfite, which converts unmethylated cytosine bases to uracil, which is complementary to adenosine in PCR primers. Two amplifications are then carried out on the bisulfite-treated DNA: one primer set anneals to DNA with cytosines (corresponding to methylated cytosine), and the other set anneals to DNA with uracil (corresponding to unmethylated cytosine). MSP used in quantitative PCR provides quantitative information about the methylation state of a given CpG island.[14]
Other modifications
Adjustments of the components in PCR is commonly used for optimal performance.
The divalent magnesium ion (Mg++) is required for PCR polymerase activity. Lower concentrations Mg++ will increase replication fidelity, while higher concentrations will introduce more mutations.[15]
Denaturants(such as DMSO) can increase amplification specificity by destabilizing non-specific primer binding. Other chemicals, such as glycerol, are stabilizers for the activity of the polymerase during amplification. Detergents (such as Triton X-100) can prevent polymerase stick to itself or to the walls of the reaction tube.
DNA polymerases occasionally incorporate mismatch bases into the extending strand. High-fidelity PCR employs enzymes with 3'-5' exonuclease activity that decreases this rate of mis-incorporation. Examples of enzymes with proofreading activity include Pfu; adjustments of the Mg++ and dNTP concentrations may help maximize the number of products that exactly match the original target DNA.
Primer modifications
Adjustments to the synthetic oligonucleotides used as primers in PCR are a rich source of modification:
Normally PCR primers are chosen from an invariant part of the genome, and might be used to amplify a polymorphic area between them. In allele-specific PCR the opposite is done. At least one of the primers is chosen from a polymorphic area, with the mutations located at (or near) its 3'-end. Under stringent conditions, a mismatched primer will not initiate replication, whereas a matched primer will. The appearance of an amplification product therefore indicates the genotype. (For more information, see SNP genotyping.)
InterSequence-Specific PCR (or ISSR-PCR) is method for DNA fingerprinting that uses primers selected from segments repeated throughout a genome to produce a unique fingerprint of amplified product lengths.[16] The use of primers from a commonly repeated segment is called Alu-PCR, and can help amplify sequences adjacent (or between) these repeats.
Primers can also be designed to be 'degenerate' – able to initiate replication from a large number of target locations. Whole genome amplification (or WGA) is a group of procedures that allow amplification to occur at many locations in an unknown genome, and which may only be available in small quantities. Other techniques use degenerate primers that are synthesized using multiple nucleotides at particular positions (the polymerase 'chooses' the correctly matched primers). Also, the primers can be synthesized with the nucleoside analog inosine, which hybridizes to three of the four normal bases. A similar technique can force PCR to perform Site-directed mutagenesis. (also see Overlap extension polymerase chain reaction)
Normally the primers used in PCR are designed to be fully complementary to the target. However, the polymerase is tolerant to mis-matches away from the 3' end. Tailed-primers include non-complementary sequences at their 5' ends. A common procedure is the use of linker-primers, which ultimately place restriction sites at the ends of the PCR products, facilitating their later insertion into cloning vectors.
An extension of the 'colony-PCR' method (above), is the use of vector primers. Target DNA fragments (or cDNA) are first inserted into a cloning vector, and a single set of primers are designed for the areas of the vector flanking the insertion site. Amplification occurs for whatever DNA has been inserted.[4]
PCR can easily be modified to produce a labeled product for subsequent use as a hybridization probe. One or both primers might be used in PCR with a radioactive or fluorescent label already attached, or labels might be added after amplification. These labeling methods can be combined with 'asymmetric-PCR' (above) to produce effective hybridization probes.
RNase H-dependent PCR (rhPCR) can reduce primer-dimer formation, and increase the number of assays in multiplex PCR. The method utilizes primers with a cleavable block on the 3’ end that is removed by the action of a thermostable RNase HII enzyme.[17]
DNA Polymerases
There are several DNA polymerases that are used in PCR.
The Klenow fragment, derived from the original DNA Polymerase I from E. coli, was the first enzyme used in PCR. Because of its lack of stability at high temperature, it needs be replenished during each cycle, and therefore is not commonly used in PCR.
The bacteriophage T4 DNA polymerase (family A) was also initially used in PCR. It has a higher fidelity of replication than the Klenow fragment, but is also destroyed by heat. T7 DNA polymerase (family B) has similar properties and purposes. It has been applied to site-directed mutagenesis[18] and Sanger sequencing.[19]
Taq polymerase, the DNA Polymerase I from Thermus aquaticus, was the first thermostable polymerase used in PCR, and is still the one most commonly used. The enzyme can be isolated from its native source, or from its cloned gene expressed in E. coli.[4] A 61kDa truncated from lacking 5'-3' exonuclease activity is known as the Stoffel fragment, and is expressed in E. coli.[20] The lack of exonuclease activity may allow it to amplify longer targets than the native enzyme. It has been commercialized as AmpliTaq and Klentaq.[21] A variant designed for hot-start PCR called the "Faststart polymerase" has also been produced. It requires strong heat activation, thereby avoiding non-specific amplification due to polymerase activity at low temperature. Many other variants have been created.[22]
Other Thermus polymerases, such as Tth polymerase I (P52028) from Thermus thermophilus, has seen some use. Tth has reverse transcriptase activity in the presence of Mn2+ ions, allowing PCR amplification from RNA targets.[23]
The archean genus Pyrococcus has proven a rich source of thermostable polymerases with proofreading activity. Pfu DNA polymerase, isolated from the P. furiosus shows a 5-fold decrease in the error rate of replication compared to Taq.[24] Since errors increase as PCR progresses, Pfu is the preferred polymerase when products are to be individually cloned for sequencing or expression. Other lesser used polymerases from this genus include Pwo (P61876) from Pyrococcus woesei, Pfx from an unnamed species, "Deep Vent" polymerase (Q51334) from strain GB-D.[25]
Vent or Tli polymerase is an extremely thermostable DNA polymerase isolated from Thermococcus litoralis. The polymerase from Thermococcus fumicolans (Tfu) has also been commercialized.[25]
Mechanism modifications
Sometimes even the basic mechanism of PCR can be modified.
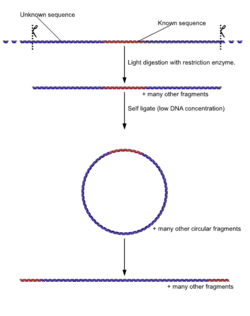
Unlike normal PCR, Inverse PCR allows amplification and sequencing of DNA that surrounds a known sequence. It involves initially subjecting the target DNA to a series of restriction enzyme digestions, and then circularizing the resulting fragments by self ligation. Primers are designed to be extended outward from the known segment, resulting in amplification of the rest of the circle. This is especially useful in identifying sequences to either side of various genomic inserts.[26]
Similarly, thermal asymmetric interlaced PCR (or TAIL-PCR) is used to isolate unknown sequences flanking a known area of the genome. Within the known sequence, TAIL-PCR uses a nested pair of primers with differing annealing temperatures. A 'degenerate' primer is used to amplify in the other direction from the unknown sequence.[27]
Isothermal amplification methods
Some DNA amplification protocols have been developed that may be used alternatively to PCR. They are isothermal, meaning that they are run at a constant temperature.[28]
Helicase-dependent amplification (HDA) is similar to traditional PCR, but uses a constant temperature rather than cycling through denaturation and annealing/extension steps. DNA Helicase, an enzyme that unwinds DNA, is used in place of thermal denaturation.[29] Loop-mediated isothermal amplification is a similar idea, but done with a strand-displacing polymerase.[30]
Nicking enzyme amplification reaction (NEAR) and its cousin strand displacement amplification (SDA) are isothermal, replicating DNA at a constant temperature using a polymerase and nicking enzyme.[28]
Recombinase Polymerase Amplification (RPA)[31] uses a recombinase to specifically pair primers with double-stranded DNA on the basis of homology, thus directing DNA synthesis from defined DNA sequences present in the sample. Presence of the target sequence initiates DNA amplification, and no thermal or chemical melting of DNA is required. The reaction progresses rapidly and results in specific DNA amplification from just a few target copies to detectable levels typically within 5–10 minutes. The entire reaction system is stable as a dried formulation and does not need refrigeration. RPA can be used to replace PCR in a variety of laboratory applications and users can design their own assays.[32]
Other types of isothermal amplification include whole genome amplification (WGA), Nucleic acid sequence-based amplification (NASBA), and transcription-mediated amplification (TMA).[28]
See also
References
- Hayden MJ, Nguyen TM, Waterman A, Chalmers KJ (2008). "Multiplex-Ready PCR: A new method for multiplexed SSR and SNP genotyping". BMC Genomics. 9: 80. doi:10.1186/1471-2164-9-80. PMC 2275739. PMID 18282271.
- Innis MA, Myambo KB, Gelfand DH, Brow MA (December 1988). "DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA". Proc. Natl. Acad. Sci. U.S.A. 85 (24): 9436–40. Bibcode:1988PNAS...85.9436I. doi:10.1073/pnas.85.24.9436. PMC 282767. PMID 3200828.
- Pierce KE, Wangh LJ (2007). "Linear-After-The-Exponential Polymerase Chain Reaction and Allied Technologies". Single Cell Diagnostics. pp. 65–85. doi:10.1007/978-1-59745-298-4_7. ISBN 978-1-58829-578-1. PMID 17876077.
{{cite book}}:|journal=ignored (help) - Saiki RK, Gelfand DH, Stoffel S, et al. (January 1988). "Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase". Science. 239 (4839): 487–91. Bibcode:1988Sci...239..487S. doi:10.1126/science.239.4839.487. PMID 2448875.
- Cheng S, Fockler C, Barnes WM, Higuchi R (June 1994). "Effective amplification of long targets from cloned inserts and human genomic DNA". Proc. Natl. Acad. Sci. U.S.A. 91 (12): 5695–9. Bibcode:1994PNAS...91.5695C. doi:10.1073/pnas.91.12.5695. PMC 44063. PMID 8202550.
- Chou Q, Russell M, Birch DE, Raymond J, Bloch W (April 1992). "Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplifications". Nucleic Acids Res. 20 (7): 1717–23. doi:10.1093/nar/20.7.1717. PMC 312262. PMID 1579465.
- Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS (July 1991). "'Touchdown' PCR to circumvent spurious priming during gene amplification". Nucleic Acids Res. 19 (14): 4008. doi:10.1093/nar/19.14.4008. PMC 328507. PMID 1861999.
- Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL (1995). "Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides". Gene. 164 (1): 49–53. doi:10.1016/0378-1119(95)00511-4. PMID 7590320.
- Pavlov AR, Pavlova NV, Kozyavkin SA, Slesarev AI (2006). "Thermostable DNA Polymerases for a Wide Spectrum of Applications: Comparison of a Robust Hybrid TopoTaq to other enzymes". In Kieleczawa J (ed.). DNA Sequencing II: Optimizing Preparation and Cleanup. Jones and Bartlett. pp. 241–257. ISBN 978-0-7637-3383-4.
- Raoult, D; G Aboudharam; E Crubezy; G Larrouy; B Ludes; M Drancourt (2000-11-07). "Molecular identification by "suicide PCR" of Yersinia pestis as the agent of medieval black death". Proc. Natl. Acad. Sci. U.S.A. 97 (23): 12800–12803. Bibcode:2000PNAS...9712800R. doi:10.1073/pnas.220225197. ISSN 0027-8424. PMC 18844. PMID 11058154.
- Androvic, Peter; Valihrach, Lukas; Elling, Julie; Sjoback, Robert; Kubista, Mikael (2017). "Two-tailed RT-qPCR: a novel method for highly accurate miRNA quantification". Nucleic Acids Research. 45 (15): e144. doi:10.1093/nar/gkx588. ISSN 0305-1048. PMC 5587787. PMID 28911110.
- Mueller PR, Wold B (November 1989). "In vivo footprinting of a muscle specific enhancer by ligation mediated PCR". Science. 246 (4931): 780–6. Bibcode:1989Sci...246..780M. doi:10.1126/science.2814500. PMID 2814500.
- Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (September 1996). "Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands". Proc. Natl. Acad. Sci. U.S.A. 93 (18): 9821–6. Bibcode:1996PNAS...93.9821H. doi:10.1073/pnas.93.18.9821. PMC 38513. PMID 8790415.
- Hernández, H; Tse, MY; Pang, SC; Arboleda, H; Forero, DA (October 2013). "Optimizing methodologies for PCR-based DNA methylation analysis". BioTechniques. 55 (4): 181–197. doi:10.2144/000114087. PMID 24107250.
- Markoulatos, P; Siafakas, N; Moncany, M (2002). "Multiplex polymerase chain reaction: a practical approach". Journal of Clinical Laboratory Analysis. 16 (1): 47–51. doi:10.1002/jcla.2058. PMC 6808141. PMID 11835531.
- E. Zietkiewicz; A. Rafalski & D. Labuda (1994). "Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification". Genomics. 20 (2): 176–83. doi:10.1006/geno.1994.1151. PMID 8020964. S2CID 41802285.
- Dobosy JR, Rose SD, Beltz KR, Rupp SM, Powers KM, Behlke MA, Walder JA (August 2011). "RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers". BMC Biotechnology. 11: 80. doi:10.1186/1472-6750-11-80. PMC 3224242. PMID 21831278.
- Venkitaraman AR (April 1989). "Use of modified T7 DNA polymerase (sequenase version 2.0) for oligonucleotide site-directed mutagenesis". Nucleic Acids Research. 17 (8): 3314. doi:10.1093/nar/17.8.3314. PMC 317753. PMID 2726477.
- "Thermo Sequenase DNA Polymerase".
- Lawyer, F. C.; Stoffel, S.; Saiki, R. K.; Chang, S. Y.; Landre, P. A.; Abramson, R. D.; Gelfand, D. H. (1993-05-01). "High-level expression, purification, and enzymatic characterization of full-length Thermus aquaticus DNA polymerase and a truncated form deficient in 5' to 3' exonuclease activity". PCR Methods and Applications. 2 (4): 275–287. doi:10.1101/gr.2.4.275. ISSN 1054-9803. PMID 8324500.
- "Applied Biosystems - Support". www6.appliedbiosystems.com.
- Villbrandt, B; Sobek, H; Frey, B; Schomburg, D (September 2000). "Domain exchange: chimeras of Thermus aquaticus DNA polymerase, Escherichia coli DNA polymerase I and Thermotoga neapolitana DNA polymerase". Protein Engineering. 13 (9): 645–54. doi:10.1093/protein/13.9.645. PMID 11054459.
- https://www.promega.in/products/pcr/rt-pcr/tth-dna-polymerase/
- Cline J, Braman JC, Hogrefe HH (September 1996). "PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases". Nucleic Acids Res. 24 (18): 3546–51. doi:10.1093/nar/24.18.3546. PMC 146123. PMID 8836181.
- van Pelt-Verkuil E, van Belkum A, Hays JP (2008). "Taq and Other Thermostable DNA Polymerases". Principles and Technical Aspects of PCR Amplification. pp. 103–18. doi:10.1007/978-1-4020-6241-4_7. ISBN 978-1-4020-6240-7.
- Ochman H, Gerber AS, Hartl DL (1 November 1988). "Genetic Applications of an Inverse Polymerase Chain Reaction". Genetics. 120 (3): 621–3. doi:10.1093/genetics/120.3.621. PMC 1203539. PMID 2852134.
- Liu YG, Whittier RF (February 1995). "Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking". Genomics. 25 (3): 674–81. doi:10.1016/0888-7543(95)80010-J. PMID 7759102.
- "Isothermal Amplification - Application Overview". New England BioLabs, Inc. 2020. Retrieved 13 August 2020.
- Vincent M, Xu Y, Kong H (August 2004). "Helicase-dependent isothermal DNA amplification". EMBO Rep. 5 (8): 795–800. doi:10.1038/sj.embor.7400200. PMC 1249482. PMID 15247927.
- Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T (2000). "Loop-mediated isothermal amplification of DNA". Nucleic Acids Res. 28 (12): 63e–63. doi:10.1093/nar/28.12.e63. PMC 102748. PMID 10871386.
- Piepenburg O, Williams CH, Stemple DL, Armes NA (2006). "DNA Detection Using Recombination Proteins". PLOS Biol. 4 (7): e204. doi:10.1371/journal.pbio.0040204. PMC 1475771. PMID 16756388.
- Lutz S, Weber P, Focke M, Faltin B, Hoffmann J, Müller C, Mark D, Roth G, Munday P, Armes N, Piepenburg O, Zengerle R, von Stetten F (April 2010). "Microfluidic lab-on-a-foil for nucleic acid analysis based on isothermal recombinase polymerase amplification (RPA)". Lab Chip. 10 (7): 887–93. doi:10.1039/b921140c. PMID 20300675.