Long QT syndrome
Long QT syndrome (LQTS) is a condition affecting repolarization (relaxing) of the heart after a heartbeat, giving rise to an abnormally lengthy QT interval.[7] It results in an increased risk of an irregular heartbeat which can result in fainting, drowning, seizures, or sudden death.[1] These episodes can be triggered by exercise or stress.[6] Some rare forms of LQTS are associated with other symptoms and signs including deafness and periods of muscle weakness.[1]
| Long QT syndrome | |
|---|---|
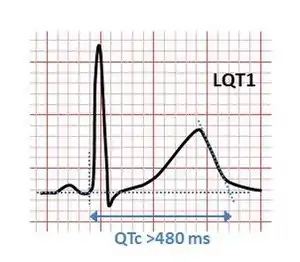 | |
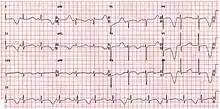
| ECG showing typical pattern of inherited Long QT syndrome (LQT1). A QT interval of >480 ms is considered abnormally long. | |
| Specialty | Cardiology |
| Symptoms | Fainting, hearing loss, seizures[1] |
| Complications | Sudden death[1] |
| Causes | Genetic, certain medications, low blood potassium, low blood calcium, heart failure[2] |
| Risk factors | Family history of sudden death[3] |
| Diagnostic method | Electrocardiogram (EKG), clinical findings, genetic testing [4][5] |
| Differential diagnosis | Brugada syndrome, arrhythmogenic right ventricular dysplasia[3] |
| Treatment | Avoiding strenuous exercise, getting sufficient potassium, beta blockers, implantable cardiac defibrillator[6] |
| Frequency | ≈ 1 in 7,000[6] |
| Deaths | ≈3,500 a year (U.S.)[6] |
Long QT syndrome may be present at birth or develop later in life.[1] The inherited form may occur by itself or as part of larger genetic disorder.[1] Onset later in life may result from certain medications, low blood potassium, low blood calcium, or heart failure.[2] Medications that are implicated include certain antiarrhythmics, antibiotics, and antipsychotics.[2] LQTS can be diagnosed using an electrocardiogram (EKG) if a corrected QT interval of greater than 450–500 milliseconds is found, but clinical findings, other EKG features, and genetic testing may confirm the diagnosis with shorter QT intervals.[4][5][8]
Management may include avoiding strenuous exercise, getting sufficient potassium in the diet, the use of beta blockers, or an implantable cardiac defibrillator.[6] For people with LQTS who survive cardiac arrest and remain untreated, the risk of death within 15 years is greater than 50%.[9][6] With proper treatment this decreases to less than 1% over 20 years.[3]
Long QT syndrome is estimated to affect 1 in 7,000 people.[6] Females are affected more often than males.[6] Most people with the condition develop symptoms before they are 40 years old.[6] It is a relatively common cause of sudden death along with Brugada syndrome and arrhythmogenic right ventricular dysplasia.[3] In the United States it results in about 3,500 deaths a year.[6] The condition was first clearly described in 1957.[10]
Signs and symptoms
.jpg.webp)
Many people with long QT syndrome have no signs or symptoms. When symptoms occur, they are generally caused by abnormal heart rhythms (arrhythmias), most commonly a form of ventricular tachycardia called Torsades de pointes (TdP). If the arrhythmia reverts to a normal rhythm spontaneously the affected person may experience lightheadedness (known as presyncope) or faint which may be preceded by a fluttering sensation in the chest.[6] If the arrhythmia continues, the affected person may experience a cardiac arrest, which if untreated may lead to sudden death.[11] Those with LQTS may also experience seizure-like activity (non-epileptic seizure) as a result of reduced blood flow to the brain during an arrhythmia.[12][13] Epilepsy is also associated with certain types of long QT syndrome.[13]
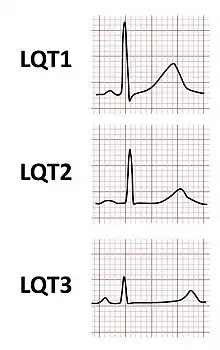
The arrhythmias that lead to faints and sudden death are more likely to occur in specific circumstances, in part determined by which genetic variant is present. While arrhythmias can occur at any time, in some forms of LQTS arrhythmias are more commonly seen in response to exercise or mental stress (LQT1), in other forms following a sudden loud noise (LQT2), and in some forms during sleep or immediately upon waking (LQT3).[11][14]
Some rare forms of long QT syndrome affect other parts of the body, leading to deafness in the Jervell and Lange-Nielsen form of the condition, and periodic paralysis in the Andersen–Tawil (LQT7) form.[4]
Risk for arrhythmias
While those with long QT syndrome have an increased risk of developing abnormal heart rhythms, the absolute risk of arrhythmias is very variable.[15] The strongest predictor of whether someone will develop TdP is whether they have experienced this arrhythmia or another form of cardiac arrest in the past.[16] Those with LQTS who have experienced syncope without an ECG having been recorded at the time are also at higher risk, as syncope in these cases is frequently due to an undocumented self-terminating arrhythmia.[16]
In addition to a history of arrhythmias, the extent to which the QT is prolonged predicts risk.[17][18] While some have QT intervals that are very prolonged, others have only slight QT prolongation, or even a normal QT interval at rest (concealed LQTS). Those with the longest QT intervals are more likely to experience TdP, and a corrected QT interval of greater than 500 ms is thought to represent those at higher risk.[19] Despite this, those with only subtle QT prolongation or concealed LQTS still have some risk of arrhythmias.[11] Overall, every 10 ms increase in the corrected QT interval is associated with a 15% increase in arrhythmic risk.[18]
As the QT prolonging effects of both genetic variants and acquired causes of LQTS are additive, those with inherited LQTS are more likely to experience TdP if given QT prolonging drugs or if they experience electrolyte problems such as low blood levels of low potassium (hypokalaemia). Similarly, those taking QT prolonging medications are more likely to experience TdP if they have a genetic tendency to a prolonged QT interval, even it this tendency is concealed.[15] Arrhythmias occur more commonly in drug-induced LQTS if the medication in question has been rapidly given intravenously, or if high concentrations of the drug are present in the person's blood.[19] The risk of arrhythmias is also higher if the person receiving the drug has heart failure, is taking digitalis, or has recently been cardioverted from atrial fibrillation.[19] Other risk factors for developing torsades de pointes among those with LQTS include female sex, increasing age, pre-existing cardiovascular disease, and abnormal liver or kidney function.[20]
Causes
There are several subtypes of long QT syndrome. These can be broadly split into those caused by genetic mutations which those affected are born with, carry throughout their lives, and can pass on to their children (inherited or congenital long QT syndrome), and those caused by other factors which cannot be passed on and are often reversible (acquired long QT syndrome).
Inherited
Inherited, or congenital long QT syndrome, is caused by genetic abnormalities. LQTS can arise from variants in several genes, leading in some cases to quite different features.[21] The common thread linking these variants is that they affect one or more ion currents leading to prolongation of the ventricular action potential, thus lengthening the QT interval.[7] Classification systems have been proposed to distinguish between subtypes of the condition based on the clinical features (and named after those who first described the condition) and subdivided by the underlying genetic variant.[22] The most common of these, accounting for 99% of cases, is Romano–Ward syndrome (genetically LQT1-6 and LQT9-16), an autosomal dominant form in which the electrical activity of the heart is affected without involving other organs.[11] A less commonly seen form is Jervell and Lange-Nielsen syndrome, an autosomal recessive form of LQTS combining a prolonged QT interval with congenital deafness.[23] Other rare forms include Andersen–Tawil syndrome (LQT7) with features including a prolonged QT interval, periodic paralysis, and abnormalities of the face and skeleton; and Timothy syndrome (LQT8) in which a prolonged QT interval is associated with abnormalities in the structure of the heart and autism spectrum disorder.[4]
Romano–Ward syndrome
LQT1 is the most common subtype of Romano–Ward syndrome, responsible for 30 to 35% of all cases.[24] The gene responsible, KCNQ1, has been isolated to chromosome 11p15.5 and encodes the alpha subunit of the KvLQT1 potassium channel. This subunit interacts with other proteins (in particular, the minK beta subunit) to create the channel, which carries the delayed potassium rectifier current IKs responsible for the repolarisation phase of the cardiac action potential.[24] Variants in KCNQ1 that decrease IKs (loss of function variants) slow the repolarisation of the action potential. This causes the LQT1 subtype of Romano–Ward syndrome when a single copy of the variant is inherited (heterozygous, autosomal dominant inheritance). Inheriting two copies of the variant (homozygous, autosomal recessive inheritance) leads to the more severe Jervell and Lange–Nielsen syndrome.[24] Conversely, variants in KCNQ1 that increase IKs lead to more rapid repolarisation and the short QT syndrome.[25]
The LQT2 subtype is the second-most common form of Romano–Ward syndrome, responsible for 25 to 30% of all cases.[24] It is caused by variants in the KCNH2 gene (also known as hERG) on chromosome 7 which encodes the potassium channel that carries the rapid inward rectifier current IKr.[24] This current contributes to the terminal repolarisation phase of the cardiac action potential, and therefore the length of the QT interval.[24]
The LQT3 subtype of Romano–Ward syndrome is caused by variants in the SCN5A gene located on chromosome 3p21–24. SCN5A encodes the alpha subunit of the cardiac sodium channel, NaV1.5, responsible for the sodium current INa which depolarises cardiac cells at the start of the action potential.[24] Cardiac sodium channels normally inactivate rapidly, but the mutations involved in LQT3 slow their inactivation leading to a small sustained 'late' sodium current. This continued inward current prolongs the action potential and thereby the QT interval.[24] While some variants in SCN5A cause LQT3, other variants can cause quite different conditions. Variants causing a reduction in the early peak current can cause Brugada syndrome and cardiac conduction disease, while other variants have been associated with dilated cardiomyopathy. Some variants which affect both the early and late sodium current can cause overlap syndromes which combine aspects of both LQT3 and Brugada syndrome.[11]
Rare Romano–Ward subtypes (LQT4-6 and LQT9-16)
LQT5 is caused by variants in the KCNE1 gene responsible for the potassium channel beta subunit MinK. This subunit, in conjunction with the alpha subunit encoded by KCNQ1, is responsible for the potassium current IKs which is decreased in LQTS.[24] LQT6 is caused by variants in the KCNE2 gene responsible for the potassium channel beta subunit MiRP1 which generates the potassium current IKr.[24] Variants that decrease this current have been associated with prolongation of the QT interval.[23] However, subsequent evidence such as the relatively common finding of variants in the gene in those without long QT syndrome, and the general need for a second stressor such as hypokalaemia to be present to reveal the QT prolongation, has suggested that this gene instead represents a modifier to susceptibility to QT prolongation.[22] Some therefore dispute whether variants in KCNE2 are sufficient to cause Romano-Ward syndrome by themselves.[22]
LQT9 is caused by variants in the membrane structural protein, caveolin-3.[24] Caveolins form specific membrane domains called caveolae in which voltage-gated sodium channels sit. Similar to LQT3, these caveolin variants increase the late sustained sodium current, which impairs cellular repolarization.[24]
LQT10 is an extremely rare subtype, caused by variants in the SCN4B gene. The product of this gene is an auxiliary beta-subunit (NaVβ4) forming cardiac sodium channels, variants in which increase the late sustained sodium current.[24] LQT13 is caused by variants in GIRK4, [26] a protein involved in the parasympathetic modulation of the heart.[24] Clinically, the patients are characterized by only modest QT prolongation, but an increased propensity for atrial arrhythmias. LQT14, LQT15 and LQT16 are caused by variants in the genes responsible for calmodulin (CALM1, CALM2, and CALM3 respectively).[24] Calmodulin interacts with several ion channels and its roles include modulation of the L-type calcium current in response to calcium concentrations, and trafficking the proteins produced by KCNQ1 and thereby influencing potassium currents.[24] The precise mechanisms by which means these genetic variants prolong the QT interval remain uncertain.[24]
Jervell and Lange–Nielsen syndrome
Jervell and Lange–Nielsen syndrome (JLNS) is a rare form of LQTS inherited in an autosomal recessive manner. In addition to severe prolongation of the QT interval, those affected are born with severe sensorineural deafness affecting both ears. The syndrome is caused by inheriting two copies of certain variant in the KCNE1 or KCNQ1 genes. The same genetic variants lead to the LQT1 and LQT5 forms of Romano-Ward syndrome if only a single copy of the variant is inherited.[11] JLNS is generally associated with a higher risk of arrhythmias than most other forms of LQTS.[4]
Andersen–Tawil syndrome (LQT7)
LQT7, also known as Andersen–Tawil syndrome, is characterised by a triad of features – in addition to a prolonged QT interval, those affected may experience intermittent weakness often occurring at times when blood potassium concentrations are low (hypokalaemic periodic paralysis), and characteristic facial and skeletal abnormalities such as a small lower jaw (micrognathia), low set ears, and fused or abnormally angled fingers and toes (syndactyly and clinodactyly).[27] The condition is inherited in an autosomal-dominant manner and is caused by mutations in the KCNJ2 gene which encodes the potassium channel protein Kir2.1.[28]
Timothy syndrome (LQT8)
LQT8, also known as Timothy syndrome combines a prolonged QT interval with fused fingers or toes (syndactyly). Abnormalities of the structure of the heart are commonly seen including ventricular septal defect, tetralogy of Fallot, and hypertrophic cardiomyopathy.[29][30] The condition presents early in life and the average life expectancy is 2.5 years with death most commonly caused by ventricular arrhythmias. Many children with Timothy syndrome who survive longer than this have features of autism spectrum disorder. Timothy syndrome is caused by variants in the calcium channel Cav1.2 encoded by the gene CACNA1c.[31]
Table of associated genes
The following is a list of genes associated with Long QT syndrome:
| Type | OMIM | Gene | Notes |
| LQT1 | 192500 | KCNQ1 | Encodes the α-subunit of the slow delayed rectifier potassium channel KV7.1 carrying the potassium current IKs.[22] |
| LQT2 | 152427 | KCNH2 | Also known as hERG. Encodes the α-subunit of the rapid delayed rectifier potassium channel KV11.1 carrying the potassium current IKr.[22] |
| LQT3 | 603830 | SCN5A | Encodes the α-subunit of the cardiac sodium channel NaV1.5 carrying the sodium current INa.[22] |
| LQT4 | 600919 | ANK2 | Encodes Ankyrin B which anchors the ion channels in the cell. Disputed whether truly disease causing versus minor QT susceptibility gene.[22] |
| LQT5 | 176261 | KCNE1 | Encodes MinK, a potassium channel β-subunit. Heterozygous inheritance causes Romano–Ward, homozygous inheritance causes Jervell and Lange–Nielsen syndrome.[22] |
| LQT6 | 603796 | KCNE2 | Encodes MiRP1, a potassium channel β-subunit. Disputed whether truly disease causing versus minor QT susceptibility gene.[22] |
| LQT7 | 170390 | KCNJ2 | Encodes inward rectifying potassium current Kir2.1 carrying the potassium current IK1. Causes Andersen–Tawil syndrome.[22] |
| LQT8 | 601005 | CACNA1c | Encodes the α-subunit CaV1.2 of the calcium channel Cav1.2 carrying the calcium current ICa(L). Causes Timothy syndrome.[22] |
| LQT9 | 611818 | CAV3 | Encodes Caveolin-3, responsible for forming membrane pouches known as caveolae. Mutations in this gene may increase the late sodium current INa.[22] |
| LQT10 | 611819 | SCN4B | Encodes the β4-subunit of the cardiac sodium channel.[22] |
| LQT11 | 611820 | AKAP9 | Encodes A-kinase associated protein which interacts with KV7.1.[22] |
| LQT12 | 601017 | SNTA1 | Encodes syntrophin-α1. Mutations in this gene may increase the late sodium current INa.[22] |
| LQT13 | 600734 | KCNJ5 | Also known as GIRK4, encodes G protein-sensitive inwardly rectifying potassium channels (Kir3.4) which carry the potassium current IK(ACh).[22] [26] |
| LQT14 | 616247 | CALM1 | Encodes calmodulin-1, a calcium-binding messenger protein that interacts with the calcium current ICa(L).[22] |
| LQT15 | 616249 | CALM2 | Encodes calmodulin-2, a calcium-binding messenger protein that interacts with the calcium current ICa(L).[22] |
| LQT16 | 114183 | CALM3 | Encodes calmodulin-3, a calcium-binding messenger protein that interacts with the calcium current ICa(L).[22] |
Acquired
Although long QT syndrome is often a genetic condition, a prolonged QT interval associated with an increased risk of abnormal heart rhythms can also occur in people without a genetic abnormality, commonly due to a side effect of medications. Drug-induced QT prolongation is often a result of treatment by antiarrhythmic drugs such as amiodarone and sotalol, antibiotics such as erythromycin, or antihistamines such as terfenadine.[20] Other drugs which prolong the QT interval include some antipsychotics such as haloperidol and ziprasidone, and the antidepressant citalopram.[32][19] Lists of medications associated with prolongation of the QT interval such as the CredibleMeds database can be found online.[33]
Other causes of acquired LQTS include abnormally low levels of potassium (hypokalaemia) or magnesium (hypomagnesaemia) within the blood. This can be exacerbated following a sudden reduction in the blood supply to the heart (myocardial infarction), low levels of thyroid hormone (hypothyroidism), and a slow heart rate (bradycardia).[34]
Anorexia nervosa has been associated with sudden death, possibly due to QT prolongation. The malnutrition seen in this condition can sometimes affect the blood concentration of salts such as potassium, potentially leading to acquired long QT syndrome, in turn causing sudden cardiac death. The malnutrition and associated changes in salt balance develop over a prolonged period of time, and rapid refeeding may further disturb the salt imbalances, increasing the risk of arrhythmias. Care must therefore be taken to monitor electrolyte levels to avoid the complications of refeeding syndrome.[35]
Factors which prolong the QT interval are additive, meaning that a combination of factors (such as taking a QT-prolonging drug and having low levels of potassium) can cause a greater degree of QT prolongation than each factor alone. This also applies to some genetic variants which by themselves only minimally prolong the QT interval but can make people more susceptible to significant drug-induced QT prolongation.[34]
Mechanisms
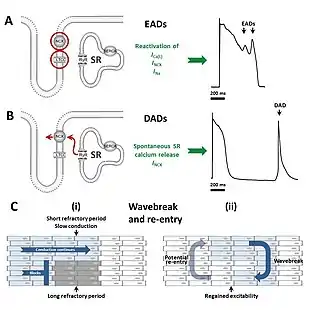
The various forms of long QT syndrome, both congenital and acquired, produce abnormal heart rhythms (arrhythmias) by influencing the electrical signals used to coordinate individual heart cells. The common theme is a prolongation of the cardiac action potential – the characteristic pattern of voltage changes across the cell membrane that occur with each heart beat.[11] Heart cells when relaxed normally have fewer positively charged ions on the inner side of their cell membrane than on the outer side, referred to as the membrane being polarised. When heart cells contract, positively charged ions such as sodium and calcium enter the cell, equalising or reversing this polarity, or depolarising the cell. After a contraction has taken place, the cell restores its polarity (or repolarises) by allowing positively charged ions such as potassium to leave the cell, restoring the membrane to its relaxed, polarised state. In long QT syndrome it takes longer for this repolarisation to occur, shown in individual cells as a longer action potential while being marked on the surface ECG as a long QT interval.[11]
The prolonged action potentials can lead to arrhythmias through several mechanisms. The arrhythmia characteristic of long QT syndrome, Torsades de Pointes, starts when an initial action potential triggers further abnormal action potentials in the form of afterdepolarisations. Early afterdepolarisations, occurring before the cell has fully repolarised, are particularly likely to be seen when action potentials are prolonged, and arise due to reactivation of calcium and sodium channels that would normally switch off until the next heartbeat is due.[36] Under the right conditions, reactivation of these currents, facilitated by the sodium-calcium exchanger, can cause further depolarisation of the cell.[36] The early afterdepolarisations triggering arrhythmias in long QT syndrome tend to arise from the Purkinje fibres of the cardiac conduction system.[37] Early afterdepolarisations may occur as single events, but may occur repeatedly leading to multiple rapid activations of the cell.[36]
Some research suggests that delayed afterdepolarisations, occurring after repolarisation has completed, may also play a role in long QT syndrome.[37] This form of afterdepolarisation originates from the spontaneous release of calcium from the intracellular calcium store known as the sarcoplasmic reticulum, forcing calcium out of cell through the sodium calcium exchanger in exchange for sodium, generating a net inward current.[36]
While there is strong evidence that the trigger for Torsades de Pointes comes from afterdepolarisations, it is less certain what sustains this arrhythmia. Some lines of evidence suggest that repeated afterdepolarisations from many sources contribute to the continuing arrhythmia.[37] However, some suggest that the arrhythmia sustains through a mechanism known as re-entry. According to this model, the action potential prolongation occurs to a variable extent in different layers of the heart muscle with longer action potentials in some layers than others.[37] In response to a triggering impulse, the waves of depolarisation will spread through regions with shorter action potentials but block in regions with longer action potentials. This allows the depolarising wavefront to bend around areas of block, potentially forming a complete loop and self-perpetuating. The twisting pattern on the ECG can be explained by movement of the core of the re-entrant circuit in the form of a meandering spiral wave.[37]
Diagnosis
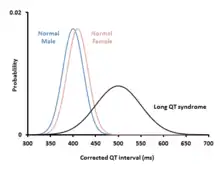
Diagnosing long QT syndrome is challenging. Whilst the hallmark of LQTS is prolongation of the QT interval, the QT interval is highly variable among both those who are healthy and those who have LQTS. This leads to overlap between the QT intervals of those with and without LQTS. 2.5% of those with genetically proven LQTS have a QT interval within the normal range.[23] Conversely, given the normal distribution of QT intervals, a proportion of healthy people will have a longer QT interval than any arbitrary cutoff.[23] Other factors beyond the QT interval should therefore be taken into account when making a diagnosis, some of which have been incorporated into scoring systems.[4]
Electrocardiogram
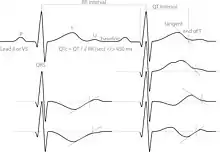
Long QT syndrome is principally diagnosed by measuring the QT interval corrected for heart rate (QTc) on a 12-lead electrocardiogram (ECG). Long QT syndrome is associated with a prolonged QTc, although in some genetically proven cases of LQTS this prolongation can be hidden, known as concealed LQTS.[23] The QTc is less than 450 ms in 95% of normal males, and less than 460 ms in 95% of normal females. LQTS is suggested if the QTc is longer than these cutoffs. However, as 5% of normal people also fall into this category, some suggest cutoffs of 470 and 480 ms for males and females respectively, corresponding with the 99th centiles of normal values.[23]
The major subtypes of inherited LQTS are associated with specific ECG features. LQT1 is typically associated with broad-based T-waves, whereas the T-waves in LQT2 are notched and of lower amplitude, whilst in LQT3 the T-waves are often late onset, being preceded by a long isoelectric segment.[23]
Schwartz score
The Schwartz score has been proposed as a method of combining clinical and ECG factors to assess how likely an individual is to have an inherited form of LQTS.[7] The table below lists the criteria used to calculate the score.
| Corrected QT interval (QTc) | ≥ 480 ms | 3 points | QTc defined according to Bazett's correction |
| 460–470 ms | 2 points | ||
| 450 ms and male gender | 1 point | ||
| Torsades de pointes | 2 points | ||
| T-wave alternans | 1 point | ||
| Notched T-waves in at least 3 leads | 1 point | ||
| Low heart rate for age (children) | 0.5 points | ||
| Syncope | with stress | 2 points | Cannot receive points both for syncope and Torsades |
| without stress | 1 point | ||
| Congenital deafness | 0.5 points | ||
| Family history | Other family member with confirmed LQTS | 1 point | Same family member cannot be counted for LQTS and sudden death |
| Sudden cardiac death in immediate family member aged <30 | 0.5 point | ||
| Score: 0–1 : low probability of LQTS; 2–3 : intermediate probability of LQTS; ≥ 4 : high probability of LQTS | |||
Other investigations
In cases of diagnostic uncertainty, other investigations may be helpful to unmask a prolonged QT. In addition to prolonging the resting QT interval, LQTS may affect how the QT changes in response to exercise and stimulation by catecholamines such as adrenaline. Provocation tests, in the form of exercise tolerance tests or direct infusion of adrenaline, can be used to detect these abnormal responses.[39] These investigations are most useful for identifying those with concealed congenital Type 1 LQTS 1 (LQT1) who have a normal QT interval at rest. While in healthy persons the QT interval shortens during exercise, in those with concealed LQT1 exercise or adrenaline infusion may lead to paradoxical prolongation of the QT interval, revealing the underlying condition.[23]
Guideline cutoffs
International consensus guidelines differ on the degree of QT prolongation required to diagnose LQTS. The European Society of Cardiology recommends that, with or without symptoms or other investigations, LQTS can be diagnosed if the corrected QT interval is longer than 480ms. They recommend that a diagnosis can be considered in the presence of a QTc of greater than 460 ms if unexplained syncope has occurred.[4] The Heart Rhythm Society guidelines are more stringent, recommending QTc cutoff of greater than 500 ms in the absence of other factors that prolong the QT, or greater than 480 ms with syncope.[5] Both sets of guidelines agree that LQTS can also be diagnosed if an individual has a Schwartz score of greater than 3 or if a pathogenic genetic variant associated with LQTS is identified, regardless of QT interval.[4][5]
Treatment
Those diagnosed with LQTS are usually advised to avoid drugs that can prolong the QT interval further or lower the threshold for TDP, lists of which can be found in public access online databases.[40] In addition to this, two intervention options are known for individuals with LQTS: arrhythmia prevention and arrhythmia termination.
Arrhythmia prevention
Arrhythmia suppression involves the use of medications or surgical procedures that attack the underlying cause of the arrhythmias associated with LQTS. Since the cause of arrhythmias in LQTS is early afterdepolarizations (EADs), and they are increased in states of adrenergic stimulation, steps can be taken to blunt adrenergic stimulation in these individuals. These include administration of beta receptor blocking agents, which decreases the risk of stress-induced arrhythmias. Nadolol, a powerful non-selective beta blocker, has been shown to reduce the arrhythmic risk in all three main genotypes (LQT1, LQT2, and LQT3).[18]
Genotype and QT interval duration are independent predictors of recurrence of life-threatening events during beta-blocker therapy.[18] To be specific, the presence of QTc >500 ms and LQT2 and LQT3 genotype are associated with the highest incidence of recurrence.[4][18] In these patients, primary prevention with use of implantable cardioverter-defibrillators can be considered.[7]
- Sodium channel blocking drugs such as mexiletine have been used to prevent arrhythmias in long QT syndrome.[41] While the most compelling indication is for those whose long QT syndrome is caused by defective sodium channels producing a sustained late current (LQT3),[41] mexiletine also shortens the QT interval in other forms of long QT syndrome including LQT1, LQT2 and LQT8.[42] As the predominant action of mexiletine is on the early peak sodium current, there are theoretical reasons why drugs which preferentially suppress the late sodium current such as ranolazine may be more effective, although evidence that this is the case in practice is limited.[42]
- Amputation of the cervical sympathetic chain (left stellectomy). This therapy is typically reserved for LQTS caused by JLNS,[7] but may be used as an add-on therapy to beta blockers in certain cases. In most cases, modern therapy favors ICD implantation if beta blocker therapy fails.
- In patients considered at high risk of life-threatening arrhythmic events,[18][43] ICD implantation may be considered as a preventive step.[4]
Arrhythmia termination
Arrhythmia termination involves stopping a life-threatening arrhythmia once it has already occurred. One effective form of arrhythmia termination in individuals with LQTS is placement of an implantable cardioverter-defibrillator (ICD). Also, external defibrillation can be used to restore sinus rhythm. ICDs are commonly used in patients with fainting episodes despite beta blocker therapy, and in patients having experienced a cardiac arrest.[4] As mentioned earlier, ICDs may be used also in patients considered at high risk of life-threatening arrhythmic events.[4][18][43]
With better knowledge of the genetics underlying LQTS, more precise treatments hopefully will become available.[44]
Outcomes
Genotype and QT interval duration are the strongest predictors of outcome for patients with LQTS.[17][18] These have been combined to create an externally validated risk score calculator, estimating a 5-year risk for each individual patient, which can help physicians to identify patients at the highest risk of life-threatening arrhythmic events.[43]
For people who experience cardiac arrest or fainting caused by LQTS and who are untreated, the risk of death within 15 years is around 50%.[9] With careful treatment this decreases to less than 1% over 20 years.[3] Those who exhibit symptoms before the age of 18 are more likely to experience a cardiac arrest.[23][45]
Epidemiology
Inherited LQTS is estimated to affect between one in 2,500 and 7,000 people.[7]
History
The first documented case of LQTS was described in Leipzig by Meissner in 1856, when a deaf girl died after her teacher yelled at her. Soon after being notified, the girl's parents reported that her older brother, also deaf, had previously died after a terrible fright.[46] This was several decades before the ECG was invented, but is likely the first described case of Jervell and Lange-Nielsen syndrome. In 1957, the first case documented by ECG was described by Anton Jervell and Fred Lange-Nielsen, working in Tønsberg, Norway.[47] Italian pediatrician Cesarino Romano, in 1963,[48] and Irish pediatrician Owen Conor Ward, in 1964,[49] separately described the more common variant of LQTS with normal hearing, later called Romano-Ward syndrome. The establishment of the International Long-QT Syndrome Registry in 1979 allowed numerous pedigrees to be evaluated in a comprehensive manner. This helped in detecting many of the numerous genes involved.[50] Transgenic animal models of the LQTS helped define the roles of various genes and hormones involved,[51][52] and recently experimental pharmacological therapies to normalize the abnormal repolarization in animals were published.[53][54]
References
- "Long QT syndrome". Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. 2017. Retrieved 14 December 2017.
- Morita H, Wu J, Zipes DP (August 2008). "The QT syndromes: long and short". Lancet. 372 (9640): 750–63. doi:10.1016/S0140-6736(08)61307-0. PMID 18761222. S2CID 41181673.
- Ferri FF (2016). Ferri's Clinical Advisor 2017 E-Book: 5 Books in 1. Elsevier Health Sciences. p. 736. ISBN 9780323448383.
- Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. (November 2015). "2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC)". Europace. 17 (11): 1601–87. doi:10.1093/europace/euv319. PMID 26318695.
- Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. (October 2013). "Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes". Europace. 15 (10): 1389–406. doi:10.1093/europace/eut272. PMID 23994779.
- "Long QT Syndrome". NHLBI, NIH. Retrieved 14 December 2017.
- Levine E, Rosero SZ, Budzikowski AS, Moss AJ, Zareba W, Daubert JP (August 2008). "Congenital long QT syndrome: considerations for primary care physicians". Cleveland Clinic Journal of Medicine. 75 (8): 591–600. doi:10.3949/ccjm.75.8.591. PMID 18756841. S2CID 4237579.
- Afzal, Muhammad Adil; Khalid, Noman; Abdullah, Muhammad; ul-Haiy, Ata; Michael, Patrick (2023). "Hydroxyzine-Induced Torsade De Pointes: A Case Report and a Literature Review". Cureus. 15 (7): e41588. doi:10.7759/cureus.41588. ISSN 2168-8184. PMC 10407684. PMID 37559846.
- Ackerman MJ, Priori SG, Dubin AM, Kowey P, Linker NJ, Slotwiner D, et al. (January 2017). "Beta-blocker therapy for long QT syndrome and catecholaminergic polymorphic ventricular tachycardia: Are all beta-blockers equivalent?". Heart Rhythm. 14 (1): e41–e44. doi:10.1016/j.hrthm.2016.09.012. PMID 27659101.
Among patients who have experienced a LQTS-triggered cardiac event (arrhythmic syncope, arrhythmic syncope followed by seizures, or aborted cardiac arrest), the untreated natural history is grim, with >50% mortality at 15 years.

- Vincent J, Abraham E, Kochanek P, Moore FA, Fink MP (2011). Textbook of Critical Care E-Book. Elsevier Health Sciences. p. 578. ISBN 978-1437715682.
- Tester DJ, Schwartz PJ, Ackerman MJ (2013). "Congenital Long QT Syndrome". In Gussak I, Antzelevitch C (eds.). Electrical Diseases of the Heart. London: Springer. pp. 439–468. doi:10.1007/978-1-4471-4881-4_27. ISBN 978-1-4471-4881-4.
- McMillan JA, Feigin RD, DeAngelis C, Jones MD (2006). Oski's Pediatrics: Principles & Practice. Lippincott Williams & Wilkins. p. 1677. ISBN 978-0-7817-3894-1.
- Madan N, Carvalho KS (February 2017). "Neurological Complications of Cardiac Disease". Seminars in Pediatric Neurology. 24 (1): 3–13. doi:10.1016/j.spen.2017.01.001. PMID 28779863.
Syncope can lead to convulsions and can easily be confused with epileptic seizures.
- Nakajima T, Kaneko Y, Kurabayashi M (2015). "Unveiling specific triggers and precipitating factors for fatal cardiac events in inherited arrhythmia syndromes". Circulation Journal. 79 (6): 1185–92. doi:10.1253/circj.CJ-15-0322. PMID 25925977.
- Trinkley KE, Page RL, Lien H, Yamanouye K, Tisdale JE (December 2013). "QT interval prolongation and the risk of torsades de pointes: essentials for clinicians". Current Medical Research and Opinion. 29 (12): 1719–26. doi:10.1185/03007995.2013.840568. PMID 24020938. S2CID 206967580.
- Barsheshet A, Dotsenko O, Goldenberg I (November 2013). "Genotype-specific risk stratification and management of patients with long QT syndrome". Annals of Noninvasive Electrocardiology. 18 (6): 499–509. doi:10.1111/anec.12117. PMC 6932574. PMID 24206565.
- Priori, Silvia G.; Schwartz, Peter J.; Napolitano, Carlo; Bloise, Raffaella; Ronchetti, Elena; Grillo, Massimiliano; Vicentini, Alessandro; Spazzolini, Carla; Nastoli, Janni; Bottelli, Georgia; Folli, Roberta (2003-05-08). "Risk Stratification in the Long-QT Syndrome". New England Journal of Medicine. 348 (19): 1866–1874. doi:10.1056/NEJMoa022147. ISSN 0028-4793. PMID 12736279.
- Mazzanti, Andrea; Maragna, Riccardo; Vacanti, Gaetano; Monteforte, Nicola; Bloise, Raffaella; Marino, Maira; Braghieri, Lorenzo; Gambelli, Patrick; Memmi, Mirella; Pagan, Eleonora; Morini, Massimo (April 2018). "Interplay Between Genetic Substrate, QTc Duration, and Arrhythmia Risk in Patients With Long QT Syndrome". Journal of the American College of Cardiology. 71 (15): 1663–1671. doi:10.1016/j.jacc.2018.01.078. hdl:20.500.12105/10498. PMID 29650123.
- Roden DM (March 2004). "Drug-induced prolongation of the QT interval". The New England Journal of Medicine. 350 (10): 1013–22. doi:10.1056/NEJMra032426. PMID 14999113. S2CID 15251057.
- Thomson C, Wright P (2014-10-15). "Long QT syndrome". The Pharmaceutical Journal. 293 (7833). Archived from the original on 2014-10-20. Retrieved 18 October 2014.
- Hedley PL, Jørgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, et al. (November 2009). "The genetic basis of long QT and short QT syndromes: a mutation update". Human Mutation. 30 (11): 1486–511. doi:10.1002/humu.21106. PMID 19862833. S2CID 19122696.
- Giudicessi JR, Wilde AA, Ackerman MJ (October 2018). "The genetic architecture of long QT syndrome: A critical reappraisal". Trends in Cardiovascular Medicine. 28 (7): 453–464. doi:10.1016/j.tcm.2018.03.003. PMC 6590899. PMID 29661707.
- Giudicessi JR, Ackerman MJ (October 2013). "Genotype- and phenotype-guided management of congenital long QT syndrome". Current Problems in Cardiology. 38 (10): 417–55. doi:10.1016/j.cpcardiol.2013.08.001. PMC 3940076. PMID 24093767.
- Bohnen MS, Peng G, Robey SH, Terrenoire C, Iyer V, Sampson KJ, Kass RS (January 2017). "Molecular Pathophysiology of Congenital Long QT Syndrome". Physiological Reviews. 97 (1): 89–134. doi:10.1152/physrev.00008.2016. PMC 5539372. PMID 27807201.
- Bjerregaard P (August 2018). "Diagnosis and management of short QT syndrome". Heart Rhythm. 15 (8): 1261–1267. doi:10.1016/j.hrthm.2018.02.034. PMID 29501667. S2CID 4519580.
- Wang F, Liu J, Hong L, Liang B, Graff C, Yang Y, Christiansen M, Olesen SP, Zhang L, Kanters JK (2013). "The phenotype characteristics of type 13 long QT syndrome with mutation in KCNJ5 (Kir3.4-G387R)". Heart Rhythm. 10 (10): 1500–6. doi:10.1016/j.hrthm.2013.07.022. PMID 23872692.
- Mazzanti, Andrea; Guz, Dmitri; Trancuccio, Alessandro; Pagan, Eleonora; Kukavica, Deni; Chargeishvili, Tekla; Olivetti, Natalia; Biernacka, Elżbieta Katarzyna; Sacilotto, Luciana; Sarquella-Brugada, Georgia; Campuzano, Oscar (April 2020). "Natural History and Risk Stratification in Andersen-Tawil Syndrome Type 1". Journal of the American College of Cardiology. 75 (15): 1772–1784. doi:10.1016/j.jacc.2020.02.033. PMID 32299589. S2CID 215803995.
- Nguyen HL, Pieper GH, Wilders R (December 2013). "Andersen-Tawil syndrome: clinical and molecular aspects". International Journal of Cardiology. 170 (1): 1–16. doi:10.1016/j.ijcard.2013.10.010. PMID 24383070.
- Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al. (February 2006). "Timothy Syndrome". GeneReviews. Seattle (WA): University of Washington, Seattle. PMID 20301577.
- Napolitano, C.; Timothy, K. W.; Bloise, R.; Priori, S. G.; Adam, M. P.; Ardinger, H. H.; Pagon, R. A.; Wallace, S. E.; Bean LJH; Mirzaa, G.; Amemiya, A. (1993). "Timothy Syndrome". PMID 20301577.
{{cite journal}}: Cite journal requires|journal=(help) - Tristani-Firouzi M, Etheridge SP (2013). Gussak I, Antzelevitch C (eds.). Andersen–Tawil and Timothy Syndromes. pp. 561–567. doi:10.1007/978-1-4471-4881-4_32. ISBN 978-1-4471-4880-7.
{{cite book}}:|work=ignored (help) - Beach SR, Celano CM, Noseworthy PA, Januzzi JL, Huffman JC (January 2013). "QTc prolongation, torsades de pointes, and psychotropic medications". Psychosomatics. 54 (1): 1–13. doi:10.1016/j.psym.2012.11.001. PMID 23295003.
- Woosley RL, Black K, Heise CW, Romero K (February 2018). "CredibleMeds.org: What does it offer?" (PDF). Trends in Cardiovascular Medicine. 28 (2): 94–99. doi:10.1016/j.tcm.2017.07.010. hdl:10150/627826. PMID 28801207.
- El-Sherif N, Turitto G, Boutjdir M (April 2018). "Acquired long QT syndrome and torsade de pointes". Pacing and Clinical Electrophysiology. 41 (4): 414–421. doi:10.1111/pace.13296. PMID 29405316. S2CID 46795997.
- Jáuregui-Garrido B, Jáuregui-Lobera I (February 2012). "Sudden death in eating disorders". Vascular Health and Risk Management. 8: 91–8. doi:10.2147/VHRM.S28652. PMC 3292410. PMID 22393299.
- Wit AL (June 2018). "Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias". Pacing and Clinical Electrophysiology. 41 (8): 883–896. doi:10.1111/pace.13419. PMID 29920724. S2CID 49310809.
- El-Sherif N, Turitto G, Boutjdir M (May 2019). "Acquired Long QT Syndrome and Electrophysiology of Torsade de Pointes". Arrhythmia & Electrophysiology Review. 8 (2): 122–130. doi:10.15420/aer.2019.8.3. PMC 6528034. PMID 31114687.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS (August 1993). "Diagnostic criteria for the long QT syndrome. An update". Circulation. 88 (2): 782–4. doi:10.1161/01.CIR.88.2.782. PMID 8339437.
- Obeyesekere MN, Klein GJ, Modi S, Leong-Sit P, Gula LJ, Yee R, et al. (December 2011). "How to perform and interpret provocative testing for the diagnosis of Brugada syndrome, long-QT syndrome, and catecholaminergic polymorphic ventricular tachycardia". Circulation: Arrhythmia and Electrophysiology. 4 (6): 958–64. doi:10.1161/CIRCEP.111.965947. PMID 22203660.
- "QT Drug List by Risk Groups". Arizona Center for Education and Research on Therapeutics. Archived from the original on 2010-12-24. Retrieved 2010-07-04.
- Mazzanti, Andrea; Maragna, Riccardo; Faragli, Alessandro; Monteforte, Nicola; Bloise, Raffaella; Memmi, Mirella; Novelli, Valeria; Baiardi, Paola; Bagnardi, Vincenzo; Etheridge, Susan P.; Napolitano, Carlo (March 2016). "Gene-Specific Therapy With Mexiletine Reduces Arrhythmic Events in Patients With Long QT Syndrome Type 3". Journal of the American College of Cardiology. 67 (9): 1053–1058. doi:10.1016/j.jacc.2015.12.033. PMC 4773513. PMID 26940925.
- Li G, Zhang L (November 2018). "The role of mexiletine in the management of long QT syndrome". Journal of Electrocardiology. 51 (6): 1061–1065. doi:10.1016/j.jelectrocard.2018.08.035. PMID 30497731. S2CID 54167081.
- Mazzanti, Andrea; Trancuccio, Alessandro; Kukavica, Deni; Pagan, Eleonora; Wang, Meng; Mohsin, Muhammad; Peterson, Derick; Bagnardi, Vincenzo; Zareba, Wojciech; Priori, Silvia G (2022-04-05). "Independent validation and clinical implications of the risk prediction model for long QT syndrome (1-2-3-LQTS-Risk)". EP Europace. 24 (4): 614–619. doi:10.1093/europace/euab238. ISSN 1099-5129. PMID 34505884.
- Compton SJ, Lux RL, Ramsey MR, Strelich KR, Sanguinetti MC, Green LS, et al. (September 1996). "Genetically defined therapy of inherited long-QT syndrome. Correction of abnormal repolarization by potassium". Circulation. 94 (5): 1018–22. doi:10.1161/01.CIR.94.5.1018. PMID 8790040. Archived from the original on 2012-07-08. Retrieved 2008-07-21.
- "Genotype risk relationship".
- Tranebjaerg L, Bathen J, Tyson J, Bitner-Glindzicz M (September 1999). "Jervell and Lange-Nielsen syndrome: a Norwegian perspective". American Journal of Medical Genetics. 89 (3): 137–46. doi:10.1002/(SICI)1096-8628(19990924)89:3<137::AID-AJMG4>3.0.CO;2-C. PMID 10704188.
- Jervell A, Lange-Nielsen F (July 1957). "Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death". American Heart Journal. 54 (1): 59–68. doi:10.1016/0002-8703(57)90079-0. PMID 13435203.
- Romano C, Gemme G, Pongiglione R (September 1963). "[Arrythmias of the Pediatric Age. II. Syncopal Attacks Due to Paroxysmal Ventricular Fibrillation. Presentation of 1st Case in Italian Pediatric Literature]". La Clinica Pediatrica (in Italian). 45: 656–83. PMID 14158288.
- Ward OC (April 1964). "A New Familial Cardiac Syndrome in Children". Journal of the Irish Medical Association. 54: 103–6. PMID 14136838.
- Moss AJ, Schwartz PJ (March 2005). "25th anniversary of the International Long-QT Syndrome Registry: an ongoing quest to uncover the secrets of long-QT syndrome". Circulation. 111 (9): 1199–201. doi:10.1161/01.CIR.0000157069.91834.DA. PMID 15753228.
- Brunner, Michael; Peng, Xuwen; Liu, Gong Xin; Ren, Xiao-Qin; Ziv, Ohad; Choi, Bum-Rak; Mathur, Rajesh; Hajjiri, Mohammed; Odening, Katja E.; Steinberg, Eric; Folco, Eduardo J. (June 2008). "Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome". The Journal of Clinical Investigation. 118 (6): 2246–2259. doi:10.1172/JCI33578. ISSN 0021-9738. PMC 2373420. PMID 18464931.
- Hornyik, Tibor; Rieder, Marina; Castiglione, Alessandro; Major, Peter; Baczko, Istvan; Brunner, Michael; Koren, Gideon; Odening, Katja E. (10 May 2021). "Transgenic rabbit models for cardiac disease research" (PDF). British Journal of Pharmacology. 179 (5): 938–957. doi:10.1111/bph.15484. PMID 33822374. S2CID 233036129.
- Bentzen, Bo Hjorth; Bahrke, Sophia; Wu, Kezhong; Larsen, Anders Peter; Odening, Katja E; Franke, Gerlind; van´s Gravesande, Karin Storm; Biermann, Jürgen; Peng, Xuwen; Koren, Gideon; Zehender, Manfred; Bode, Christoph; Grunnet, Morten; Brunner, Michael (February 2011). "Pharmacological Activation of Kv11.1 in Transgenic Long QT-1 Rabbits". Journal of Cardiovascular Pharmacology. 57 (2): 223–230. doi:10.1097/FJC.0b013e318203a44d. PMID 21135701. S2CID 3282707.
- Castiglione, Alessandro; Hornyik, Tibor; Wülfers, Eike M; Giammarino, Lucilla; Edler, Iask; Jowais, Jessica J; Rieder, Marina; Perez-Feliz, Stefanie; Koren, Gideon; Bősze, Zsuzsanna; Varró, András; Zehender, Manfred; Brunner, Michael; Bode, Christoph; Liin, Sara I; Larsson, Hans Peter; Baczkó, István; Odening, Katja E (3 October 2021). "Docosahexaenoic acid normalizes QT interval in long QT type 2 transgenic rabbit models in a genotype-specific fashion". EP Europace. 24 (3): 511–522. doi:10.1093/europace/euab228. PMC 9125797. PMID 34601592.
{kind=link}
- Notes
- Goldman L (2011). Goldman's Cecil Medicine (24th ed.). Philadelphia: Elsevier Saunders. p. 1196. ISBN 978-1437727883.