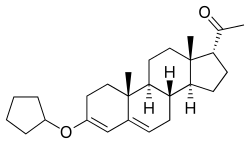
Quingestrone
Quingestrone, also known as progesterone 3-cyclopentyl enol ether (PCPE) and sold under the brand name Enol-Luteovis, is a progestin medication which was previously used in birth control pills in Italy but is now no longer marketed.[1][2][3][4][5] It is taken by mouth.[6]
 | |
| Clinical data | |
|---|---|
| Trade names | Enol-Luteovis |
| Other names | W-3399; Progesterone 3-cyclopentyl enol ether; PCPE; 3-Cyclopentyloxypregna-3,5-dien-20-one |
| Routes of administration | By mouth |
| Drug class | Progestogen; Progestin; Progestogen ether; Neurosteroid |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C26H38O2 |
| Molar mass | 382.588 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
Quingestrone is a progestin, or a synthetic progestogen, and hence is an agonist of the progesterone receptor, the biological target of progestogens like progesterone.[1][7][8] It has weak glucocorticoid activity.[9][10][11]
Quingestrone was introduced for medical use by 1962.[6][12] It is no longer available.[13]
Medical uses
Quingestrone was formerly used in combination with ethinylestradiol or mestranol in combined birth control pills in Italy.[2][3] The medication was studied in the clinical prevention of miscarriage during pregnancy, but insufficient efficacy was observed at the dosage assessed (100 mg/day orally).[14][15][16][17]
Side effects
Pharmacology
Pharmacodynamics
Along with the retroprogesterone derivative dydrogesterone, quingestrone has been described as a "true" progesterone derivative or progestogen due to its close similarity to natural progesterone.[18][12] Similarly to progesterone, dydrogesterone, and hydroxyprogesterone caproate, quingestrone is a pure progestogen and lacks any androgenic effects.[19] As such, it poses no risk of androgenic side effects or virilizing teratogenic effects on female fetuses.[19] Quingestrone is said to influence the hypothalamic–pituitary–adrenal axis similarly to progesterone and medroxyprogesterone acetate, producing adrenal suppression at sufficiently high doses, and this suggests that it possesses weak glucocorticoid activity similarly to progesterone.[9][10][11]
Quingestrone is a very weak progestogen.[8][11] When administered orally or intraperitoneally in animals, the medication showed 1/80 and 1/20 the potency of subcutaneously injected progesterone, respectively.[8] Similarly, oral doses of quingestrone of 10 to 20 times those of subcutaneous progesterone were insufficient to maintain pregnancy in animals, and oral or intraperitoneal doses of quingestrone 20 to 40 times those of oral or intraperitoneal progesterone were unable to potentiate hexobarbital-induced anesthesia in animals (which progesterone does and is thought to do by inhibiting the hepatic metabolism of barbiturates).[8] With oral administration of equal doses of progesterone and quingestrone in animals, 3 to 4 times less pregnanediol was recovered from urine with quingestrone.[8] The fact that quingestrone is more potent by intraperitoneal than oral administration in animals suggests that it is transformed into a less active metabolite in the intestines.[8]
The effective dosage of quingestrone in the menstrual delay test has been studied.[20]
Quingestrone has no anesthetic effect in animals, in contrast to progesterone.[21][22]
Pharmacokinetics
Quingestrone has been suggested to act as a prodrug of progesterone via slow hydrolysis in the body.[14][23] Indeed, it produces similar metabolites (e.g., pregnanediols and allopregnanediols) as progesterone,[14][24] although with differing ratios,[25][4] and notably is the only progestin that is known to produce pregnanediol as a metabolite.[6] Subsequent research has cast doubt on the notion that quingestrone is a prodrug of progesterone however, and indicates that it instead is directly metabolized into pregnanediols without intermediate conversion into progesterone.[8] Based on its chemical structure, quingestrone may be transformed into 3α-dihydroprogesterone and/or 3β-dihydroprogesterone and then further metabolized into pregnanolones and pregnanediols. 3β-Dihydroprogesterone has been reported to possess about the same progestogenic potency as progesterone in the Clauberg test, whereas 3α-dihydroprogesterone was not assessed.[26][27]
Relative to progesterone, quingestrone shows improved pharmacokinetics, including higher potency,[25] oral activity,[28] and a longer terminal half-life and hence duration of action.[23] This is considered to be due to its higher lipophilicity,[23] being stored into and slowly released from fat.[5][14] Quingestrone also shows slower metabolism and more stable blood levels, with a longer time to peak concentrations and a less intense peak compared to progesterone.[7] The bioavailability of quingestrone is highest when it is given as a sesame seed oil solution (compared to an oil suspension (~2-fold less) or micronization (~7-fold less)).[24]
The C3 enol ethers of progesterone are less suited for use via depot injection relative to progestogen esters like hydroxyprogesterone caproate due to their susceptibility to oxidative metabolism.[29]
The pharmacokinetics of quingestrone have been reviewed.[21]
Chemistry
Quingestrone, also known as progesterone 3-cyclopentyl enol ether (PCPE) or as 3-cyclopentyloxypregna-3,5-dien-20-one, is a synthetic pregnane steroid and a derivative of progesterone.[1] It is specifically the 3-cyclopentyl enol ether of progesterone.[1] Quingestrone is closely related to progesterone 3-acetyl enol ether and pentagestrone acetate (17α-acetoxyprogesterone 3-cyclopentyl enol ether).[1]
Synthesis
Chemical syntheses of quingestrone have been published.[21]
History
Quingestrone appears to have been first synthesized in 1936.[30] It was introduced for medical use in Italy by 1962.[6][12]
Society and culture
Generic names
Quingestrone is the generic name of the drug and its INNTooltip International Nonproprietary Name and USANTooltip United States Adopted Name.[1] It is also known by its developmental code name W-3399.[1]
References
- Elks J, Ganellin CR, eds. (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. p. 1058. doi:10.1007/978-1-4757-2085-3. ISBN 978-1-4757-2085-3. OCLC 898564124.
- International Planned Parenthood Federation. Medical Committee. Oral Advisory Group (1965). Handbook on oral contraception. Little, Brown. p. 18. OCLC 2717593.
- Larrey D, Geneve J, Pessayre D, Machayekhi JP, Degott C, Benhamou JP (February 1987). "Prolonged cholestasis after cyproheptadine-induced acute hepatitis". Journal of Clinical Gastroenterology. 9 (1): 102–104. doi:10.1097/00004836-198702000-00026. PMID 3559100.
She had been taking an oral contraceptive combining ethinylestradiol and quingestrone since 1980.
- Hawkins DF (1974). Obstetric therapeutics: clinical pharmacology and therapeutics in obstetric practice. Baillière Tindall. pp. 129, 138, 145. ISBN 978-0-7020-0471-1.
- Bentley PJ (1980). Endocrine Pharmacology: Physiological Basis and Therapeutic Applications. CUP Archive. pp. 274–. ISBN 978-0-521-22673-8.
- Current Medicine and Drugs. 1962. ISSN 0590-4048.
Enol Luteovis (3 cyclo-pentyl enol ether of progesterone) is the only oral progestin producing pregnanediol as a metabolite. It is not very potent and probably carries very little risk of producing virilizing effects on a female foetus. Thus it is more closely related to progesterone than the other synthetic progestins.
- Caie E, Klopper A (January 1964). "The Urinary Excretion of Pregnanediol After the Administration of an Oral Gestagen (Progesterone Cyclopentyl Enol Ether)". The Journal of Endocrinology. 28 (2): 221–222. CiteSeerX 10.1.1.1028.4921. doi:10.1677/joe.0.0280221. PMID 14112260.
- Meli A, Wolff A, Lucker WE, Steinetz BG (March 1965). "The Biological Profile of Progesterone 3-Cyclopentyl Enol Ether as Compared with That of Progesterone". Proceedings of the Society for Experimental Biology and Medicine. 118 (3): 714–717. doi:10.3181/00379727-118-29947. PMID 14264537. S2CID 11891451.
- Martini L (1966). Neuroendocrinology. Academic Press. p. 331. ISBN 9780124753525. LCCN 66026256.
- Steinetz BG, Beach VL, DiPasquale G, Battista Jr JV (1965). "Effects of different gestagenic steroid types on plasma-free corticosteroid levels in ACTH-treated rats". Steroids. 5 (1): 93–108. doi:10.1016/0039-128X(65)90134-0. ISSN 0039-128X.
- Gaunt R, Steinetz BG, Chart JJ (1968). "Pharmacologic alteration of steroid hormone functions". Clinical Pharmacology and Therapeutics. 9 (5): 657–681. doi:10.1002/cpt196895657. PMID 4175595. S2CID 38695246.
An interesting substance which has received little attention is the 3-cyclopentyl enol ether of progesterone (quingestrone). It is a very weak progestational agent, requiring 50 mg. per rat for pregnancy maintenance. 100 At this dose quingestrone reduced adrenal weight in male rats to the level observed after hypophysectomy and prevented any rise in plasma corticosteroids in response to a maximally stimulating dose of ACTH.H5 This strongly suggests a direct adrenal effect although the substance may in addition suppress ACTH secretion. It is doubtful, however, that progestational agents have clinically important effects on the human adrenal in the doses conventionally used. Nevertheless, in view of the prolonged exposure of women to gestogens for contraception, this factor deserves continued surveillance.
- Appleby B (February 1962). "Norethisterone in the control of menopausal symptoms". Lancet. 1 (7226): 407–409. doi:10.1016/s0140-6736(62)91363-6. PMID 13861933.
Dr. Appleby would be doing a scientific service if he extended his trial using [...] preferably, a true progesterone derivative, such as [...] progesterone cyclopentyl enol ether ('Enol Luteovis', Vister).
- http://www.micromedexsolutions.com/micromedex2/
- Burton ER, Wachtel EG (August 1967). "A clinical trial and cytological assessment of enol LUTEOVIS IN THE TREATMENT OF THREATENED AND RECURRENT ABORTION". The Journal of Obstetrics and Gynaecology of the British Commonwealth. 74 (4): 533–536. doi:10.1111/j.1471-0528.1967.tb03986.x. PMID 5340429. S2CID 31602503.
- Vitamins and Hormones. Academic Press. 9 February 1973. pp. 332–. ISBN 978-0-08-086627-7.
- Fraser IS (1998). Estrogens and Progestogens in Clinical Practice. Churchill Livingstone. ISBN 978-0-443-04706-0.
- Goldstein P, Berrier J, Rosen S, Sacks HS, Chalmers TC (March 1989). "A meta-analysis of randomized control trials of progestational agents in pregnancy". British Journal of Obstetrics and Gynaecology. 96 (3): 265–274. doi:10.1111/j.1471-0528.1989.tb02385.x. PMID 2653414. S2CID 72030836.
- Wachtel EG (1969). Exfoliative cytology in gynaecological practice. Appleton-Century-Crofts. p. 134. ISBN 9780407169012. LCCN 77008744.
- Baird D, Kerr JM (1969). Combined textbook of obstetrics and gynæcology for students and practitioners. E. & S. Livingstone. ISBN 9780443000454. LCCN 70360656.
- Edgren RA, Sturtevant FM (August 1976). "Potencies of oral contraceptives". American Journal of Obstetrics and Gynecology. 125 (8): 1029–1038. doi:10.1016/0002-9378(76)90804-8. PMID 952300.
- Junkmann K (1968). Die Gestagene. Springer-Verlag. pp. 10, 275, 524. ISBN 978-3-642-99941-3.
- Ercoli A, Gardi R (1960). "Δ4-3-Keto Steroidal Enol Ethers. Paradoxical Dependency of Their Effectiveness on the Administration Route". Journal of the American Chemical Society. 82 (3): 746–748. doi:10.1021/ja01488a062. ISSN 0002-7863.
- Charman WN, Porter CJ (1996). "Lipophilic prodrugs designed for intestinal lymphatic transport". Advanced Drug Delivery Reviews. 19 (2): 149–169. doi:10.1016/0169-409X(95)00105-G. ISSN 0169-409X.
- Fatouros DG, Karpf DM, Nielsen FS, Mullertz A (August 2007). "Clinical studies with oral lipid based formulations of poorly soluble compounds". Therapeutics and Clinical Risk Management. 3 (4): 591–604. PMC 2374933. PMID 18472981.
- "POPLINE retirement". 2019-09-06.
- Junkermann H, Runnebaum B, Lisboa BP (July 1977). "New progesterone metabolites in human myometrium". Steroids. 30 (1): 1–14. doi:10.1016/0039-128X(77)90131-3. PMID 919010. S2CID 28420255.
In the Clauberg bioassay the 3β-hydroxy-4-pregnen-20-one shows about the same potency as progesterone (34). In regard to the biological activity of the 3α epimer no data are available.
- Pincus G, Miyake T, Merrill AP, Longo P (November 1957). "The bioassay of progesterone". Endocrinology. 61 (5): 528–533. doi:10.1210/endo-61-5-528. PMID 13480263.
- De Lee JB (1965). "The ... Year Book of Obstetrics and Gynecology". Yearbook of Obstetrics and Gynecology (O). Year Book Publishers: 150. ISSN 0084-3911. LCCN cdr38000020.
- Junkmann K (1954). "Gestagens of prolonged action". Naunyn-Schmiedebergs Archiv für Pharmakologie und Experimentelle Pathologie. 223: 244–53. ISSN 0365-5423.
Among a large no. of pregnane derivs. the esters of 17-α-hydroxyprogesterone (I), itself of weak lutein hormone action, have a strong and long-lasting gestagen action. The optimal results are obtained with I caproate. It permits the administration of depot doses in clear solns. Within the range of dosage used no androgenic effect was noted. It has no influence on growth and on the secondary sex characteristics in infantile and adult castrate male rats. The 3-enol esters of progesterone, which have a somewhat prolonged action, are less suited for depot administration because of their oxidizability.
- Westphal U (1936). "Über Enolacetate des Progesterons und Testosterons". Die Naturwissenschaften. 24 (44): 696–697. Bibcode:1936NW.....24..696W. doi:10.1007/BF01491541. ISSN 0028-1042. S2CID 41442733.