TNNT1
Slow skeletal muscle troponin T (sTnT) is a protein that in humans is encoded by the TNNT1 gene.[5][6]
| TNNT1 | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | TNNT1, ANM, NEM5, STNT, TNT, TNTS, troponin T1, slow skeletal type | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 191041 MGI: 1333868 HomoloGene: 20704 GeneCards: TNNT1 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
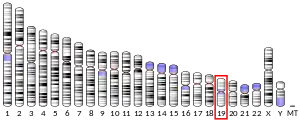




The TNNT1 gene is located at 19q13.4 in the human chromosomal genome, encoding the slow twitch skeletal muscle isoform of troponin T (ssTnT). ssTnT is an ~32-kDa protein consisting of 262 amino acids (including the first methionine) with an isoelectric point (pI) of 5.95. It is the tropomyosin binding and thin filament anchoring subunit of the troponin complex in the sarcomeres of slow twitch skeletal muscle fibers.[7][8][9] TNNT1 gene is specifically expressed in slow skeletal muscle of vertebrates, with one exception that dry land toad (Bufo) cardiac muscle expresses ssTnT other than cardiac TnT.[10]
Evolution
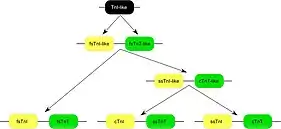
Three homologous genes have evolved in vertebrates, encoding three muscle type specific isoforms of TnT.[9] Each of the TnT isoform genes is linked to one of the three troponin I[11] isoform genes encoding the inhibitory subunit of the troponin complex, in chromosomal DNA to form three gene pairs: The fast skeletal muscle TnI (fsTnI)-fsTnT, ssTnI-cardiac (cTnT) and cTnI-ssTnT gene pairs. Sequence and epitope conservation studies suggested that genes encoding the muscle type specific TnT and TnI isoforms may have evolved from duplications of a fsTnI-like-fsTnT-like gene pair.[12] Evolutionary lineage of the three TnI-TnT gene pairs shows that cTnI-ssTnT is the newest[12] and most closely linked.[13]
Protein sequence alignment demonstrated that TNNT1 genes are highly conserved among vertebrate species (Fig. 2), especially in the middle and C-terminal regions, while the three muscle type isoforms are significantly diverged among vertebrate species.[8][9]
Alternative splicing
In mammalian and avian species, TNNT1 gene has a total of 14 exons, among which exon 5 encoding an 11-amino acid in the N-terminal region is alternatively spliced, generating a high molecular weight and a low molecular weight slow TnT splice forms (Jin, Chen et al. 1998).[14][15] Biochemical studies showed that TnT splice forms have detectable different molecular conformation in the middle and C-terminal regions, producing different binding affinities for TnI and tropomyosin.[8][9] The alternative splice forms of ssTnT play a role in skeletal muscle adaptation in physiologic and pathological conditions.[16] Alternative splicing at alternative acceptor sites of intron 5 generates a single amino acid difference in the N-terminal region of ssTnT,[15] of which functional significance has not been established.
Clinical significance
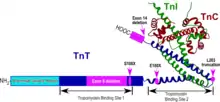
A nonsense mutation E180X in the exon 11 of TNNT1 gene causes Amish Nemaline Myopathy (ANM), which is a severe form of recessive nemaline myopathy originally found in the Old Order Amish population in Pennsylvania, USA.[17][18] Truncation of the ssTnT polypeptide chain by the E180X mutation deletes the tropomyosin-binding site 2[19] as well as the binding sites for TnI and troponin C (TnC) in the C-terminal region (Fig. 3). Consistent with the recessive phenotype, the truncated ssTnT is incapable of incorporation into the myofilaments and completely degraded in muscle cells.[20]
Tnnt1 gene targeted mouse studies reproduced the myopathic phenotypes of ANM.[21][22] ssTnT null mice showed significantly decreased type I slow fibers in diaphragm and soleus muscles with hypertrophy of type II fast fibers, increased fatigability, and active regeneration of slow fibers (Wei, Lu et al. 2014).[21]
Recent case reports identified three more mutations in TNNT1 gene to cause nemaline myopathies outside the Amish population. A nonsense mutation S108X in exon 9 was identified in a Hispanic male patient with severe recessive nemaline myopathy phenotype.[23] A Dutch patient with compound heterozygous TNNT1 gene mutations that cause exon 8 and exon 14 deletions also presents nemaline myopathy phenotypes.[24] A rearrangement in TNNT1 gene (c.574_577 delins TAGTGCTGT) leading to aberrant splicing that causes C-terminal truncation of the protein (L203 truncation) was reported in 9 Palestinian patients from 7 unrelated families with recessively inherited nemaline Myopathy.[25]
Illustrated in Fig. 3, the S108X mutation truncates ssTnT protein to cause a loss of functional structures equivalent to that of E180X. The exon 8 deletion destructs the middle region tropomyosin-binding site 1.[19][26] The L203 truncation deletes the binding sites for TnI and TnC but preserves both tropomyosin-binding sites 1 and 2.[19] It remains to be investigated whether this novel mutation is able to bind the actin-tropomyosin thin filament in vivo and how it causes recessive nemaline myopathy.
Alternative splicing of exon 5 produces high and low molecular weight splice forms of ssTnT. The low molecular ssTnT was significantly upregulated in type 1 (demyelination) but not type 2 (axon loss) Charcot-Marie-Tooth disease,[16] suggesting that structural modification of TnT in the myofilaments may contribute to adaptation to abnormalities in neuronal activation.
Notes
References
- GRCh38: Ensembl release 89: ENSG00000105048 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000064179 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Samson F, de Jong PJ, Trask BJ, Koza-Taylor P, Speer MC, Potter T, Roses AD, Gilbert JR (Aug 1992). "Assignment of the human slow skeletal troponin T gene to 19q13.4 using somatic cell hybrids and fluorescence in situ hybridization analysis". Genomics. 13 (4): 1374–5. doi:10.1016/0888-7543(92)90077-6. PMID 1505979.
- "Entrez Gene: TNNT1 troponin T type 1 (skeletal, slow)".
- Perry SV (Aug 1998). "Troponin T: genetics, properties and function". Journal of Muscle Research and Cell Motility. 19 (6): 575–602. doi:10.1023/a:1005397501968. PMID 9742444. S2CID 1882224.
- Jin JP, Zhang Z, Bautista JA (2008). "Isoform diversity, regulation, and functional adaptation of troponin and calponin". Critical Reviews in Eukaryotic Gene Expression. 18 (2): 93–124. doi:10.1615/critreveukargeneexpr.v18.i2.10. PMID 18304026.
- Wei B, Jin JP (Jan 2011). "Troponin T isoforms and posttranscriptional modifications: Evolution, regulation and function". Archives of Biochemistry and Biophysics. 505 (2): 144–54. doi:10.1016/j.abb.2010.10.013. PMC 3018564. PMID 20965144.
- Feng HZ, Chen X, Hossain MM, Jin JP (Aug 2012). "Toad heart utilizes exclusively slow skeletal muscle troponin T: an evolutionary adaptation with potential functional benefits". The Journal of Biological Chemistry. 287 (35): 29753–64. doi:10.1074/jbc.M112.373191. PMC 3436204. PMID 22778265.
- Dweck D, Sanchez-Gonzalez MA, Chang AN, Dulce RA, Badger CD, Koutnik AP, Ruiz EL, Griffin B, Liang J, Kabbaj M, Fincham FD, Hare JM, Overton JM, Pinto JR (Aug 2014). "Long term ablation of protein kinase A (PKA)-mediated cardiac troponin I phosphorylation leads to excitation-contraction uncoupling and diastolic dysfunction in a knock-in mouse model of hypertrophic cardiomyopathy". The Journal of Biological Chemistry. 289 (33): 23097–111. doi:10.1074/jbc.M114.561472. PMC 4132808. PMID 24973218.
- Chong SM, Jin JP (May 2009). "To investigate protein evolution by detecting suppressed epitope structures". Journal of Molecular Evolution. 68 (5): 448–60. Bibcode:2009JMolE..68..448C. doi:10.1007/s00239-009-9202-0. PMC 2752406. PMID 19365646.
- Huang QQ, Jin JP (Dec 1999). "Preserved close linkage between the genes encoding troponin I and troponin T, reflecting an evolution of adapter proteins coupling the Ca(2+) signaling of contractility". Journal of Molecular Evolution. 49 (6): 780–8. Bibcode:1999JMolE..49..780H. doi:10.1007/pl00006600. PMID 10594179. S2CID 9839814.
- Gahlmann R, Troutt AB, Wade RP, Gunning P, Kedes L (Nov 1987). "Alternative splicing generates variants in important functional domains of human slow skeletal troponin T". The Journal of Biological Chemistry. 262 (33): 16122–6. doi:10.1016/S0021-9258(18)47705-8. PMID 2824479.
- Huang QQ, Chen A, Jin JP (Mar 1999). "Genomic sequence and structural organization of mouse slow skeletal muscle troponin T gene". Gene. 229 (1–2): 1–10. doi:10.1016/s0378-1119(99)00051-7. PMID 10095098.
- Larsson L, Wang X, Yu F, Höök P, Borg K, Chong SM, Jin JP (Sep 2008). "Adaptation by alternative RNA splicing of slow troponin T isoforms in type 1 but not type 2 Charcot-Marie-Tooth disease". American Journal of Physiology. Cell Physiology. 295 (3): C722–31. doi:10.1152/ajpcell.00110.2008. PMC 2544436. PMID 18579801.
- Johnston JJ, Kelley RI, Crawford TO, Morton DH, Agarwala R, Koch T, Schäffer AA, Francomano CA, Biesecker LG (Oct 2000). "A novel nemaline myopathy in the Amish caused by a mutation in troponin T1". American Journal of Human Genetics. 67 (4): 814–21. doi:10.1086/303089. PMC 1287886. PMID 10952871.
- Jin JP, Brotto MA, Hossain MM, Huang QQ, Brotto LS, Nosek TM, Morton DH, Crawford TO (Jul 2003). "Truncation by Glu180 nonsense mutation results in complete loss of slow skeletal muscle troponin T in a lethal nemaline myopathy". The Journal of Biological Chemistry. 278 (28): 26159–65. doi:10.1074/jbc.M303469200. PMID 12732643.
- Jin JP, Chong SM (Aug 2010). "Localization of the two tropomyosin-binding sites of troponin T". Archives of Biochemistry and Biophysics. 500 (2): 144–50. doi:10.1016/j.abb.2010.06.001. PMC 2904419. PMID 20529660.
- Martin AF (Jan 1981). "Turnover of cardiac troponin subunits. Kinetic evidence for a precursor pool of troponin-I". The Journal of Biological Chemistry. 256 (2): 964–8. doi:10.1016/S0021-9258(19)70073-8. PMID 7451483.
- Feng HZ, Wei B, Jin JP (Nov 2009). "Deletion of a genomic segment containing the cardiac troponin I gene knocks down expression of the slow troponin T gene and impairs fatigue tolerance of diaphragm muscle". The Journal of Biological Chemistry. 284 (46): 31798–806. doi:10.1074/jbc.M109.020826. PMC 2797250. PMID 19797054.
- Wei B, Lu Y, Jin JP (Mar 2014). "Deficiency of slow skeletal muscle troponin T causes atrophy of type I slow fibres and decreases tolerance to fatigue". The Journal of Physiology. 592 (Pt 6): 1367–80. doi:10.1113/jphysiol.2013.268177. PMC 3961093. PMID 24445317.
- Marra JD, Engelstad KE, Ankala A, Tanji K, Dastgir J, De Vivo DC, Coffee B, Chiriboga CA (May 2015). "Identification of a novel nemaline myopathy-causing mutation in the troponin T1 (TNNT1) gene: a case outside of the old order Amish". Muscle & Nerve. 51 (5): 767–72. doi:10.1002/mus.24528. PMID 25430424. S2CID 29391961.
- van der Pol WL, Leijenaar JF, Spliet WG, Lavrijsen SW, Jansen NJ, Braun KP, Mulder M, Timmers-Raaijmakers B, Ratsma K, Dooijes D, van Haelst MM (Mar 2014). "Nemaline myopathy caused byTNNT1 mutations in a Dutch pedigree". Molecular Genetics & Genomic Medicine. 2 (2): 134–7. doi:10.1002/mgg3.52. PMC 3960055. PMID 24689076.
- Abdulhaq UN, Daana M, Dor T, Fellig Y, Eylon S, Schuelke M, Shaag A, Elpeleg O, Edvardson S (2015). "Nemaline body myopathy caused by a novel mutation in Troponin T1 (TNNT1)". Muscle Nerve. 53 (4): 564–9. doi:10.1002/mus.24885. PMID 26296490. S2CID 41713544.
- Amarasinghe C, Jin JP (Jun 2015). "N-Terminal Hypervariable Region of Muscle Type Isoforms of Troponin T Differentially Modulates the Affinity of Tropomyosin-Binding Site 1". Biochemistry. 54 (24): 3822–30. doi:10.1021/acs.biochem.5b00348. PMID 26024675.
- Yuasa K, Michibata H, Omori K, Yanaka N (Dec 1999). "A novel interaction of cGMP-dependent protein kinase I with troponin T". The Journal of Biological Chemistry. 274 (52): 37429–34. doi:10.1074/jbc.274.52.37429. PMID 10601315.
- Jin JP, Chen A, Huang QQ (Jul 1998). "Three alternatively spliced mouse slow skeletal muscle troponin T isoforms: conserved primary structure and regulated expression during postnatal development". Gene. 214 (1–2): 121–9. doi:10.1016/s0378-1119(98)00214-5. PMID 9651500.
- Palm T, Graboski S, Hitchcock-DeGregori SE, Greenfield NJ (Nov 2001). "Disease-causing mutations in cardiac troponin T: identification of a critical tropomyosin-binding region". Biophysical Journal. 81 (5): 2827–37. Bibcode:2001BpJ....81.2827P. doi:10.1016/S0006-3495(01)75924-3. PMC 1301748. PMID 11606294.
- Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ (Apr 2005). "Ca(2+)-regulated structural changes in troponin". Proceedings of the National Academy of Sciences of the United States of America. 102 (14): 5038–43. Bibcode:2005PNAS..102.5038V. doi:10.1073/pnas.0408882102. PMC 555973. PMID 15784741.