Craniosynostosis
Craniosynostosis is a condition in which one or more of the fibrous sutures in a young infant's skull prematurely fuses by turning into bone (ossification),[2] thereby changing the growth pattern of the skull.[3] Because the skull cannot expand perpendicular to the fused suture, it compensates by growing more in the direction parallel to the closed sutures.[3] Sometimes the resulting growth pattern provides the necessary space for the growing brain, but results in an abnormal head shape and abnormal facial features.[3] In cases in which the compensation does not effectively provide enough space for the growing brain, craniosynostosis results in increased intracranial pressure leading possibly to visual impairment, sleeping impairment, eating difficulties, or an impairment of mental development combined with a significant reduction in IQ.[4]
| Craniosynostosis | |
|---|---|
| Other names | Craniostenosis[1] |
 | |
| Child with premature closure (craniosynostosis) of the lambdoid suture. Notice the swelling on the right side of the head | |
| Specialty | Medical genetics |
| Complications | Increased intracranial pressure |
| Usual onset | during young age |
Craniosynostosis occurs in one in 2000 births. Craniosynostosis is part of a syndrome in 15% to 40% of affected patients, but it usually occurs as an isolated condition.[5][6] The term is from cranio, cranium; + syn, together; + ost, relating to bone; + osis, denoting a condition.
Signs and symptoms
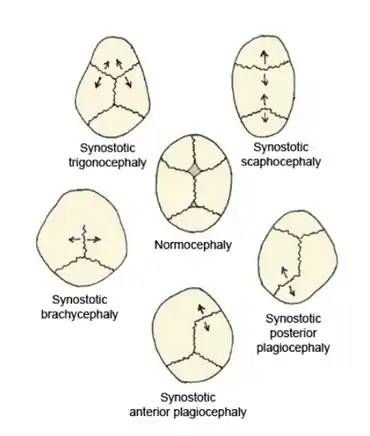
Children born with craniosynostosis have a distinct phenotype, i.e., appearance—observable traits caused by the expression of a condition's genes. The features of craniosynostosis' particular phenotype are determined by which suture is closed.[7] The fusion of this suture causes a certain change in the shape of the skull; a deformity of the skull.[7]
Virchow's law dictates that, when premature suture closure occurs, growth of the skull typically is restricted perpendicularly to the fused suture and enhanced in a plane parallel to it, thus trying to provide space for the fast-growing brain.[8] Using this law, the pattern of skull deformity in craniosynostosis often may be predicted.[8]
Scaphocephaly
An illustrative example of this phenomenon is scaphocephaly; the name providing a direct hint regarding the deformity of the skull. The literal meaning of the Greek derived word 'scaphocephaly' is boat-head. A synonymous term is 'dolichocephaly' (the prefix dolicho- means elongated).[9]
Premature sagittal suture closure restricts growth in a perpendicular plane, thus the head will not grow sideways and will remain narrow.[10][11] This is best seen in a view standing above the child looking downward at the top of the head.[12] Compensatory growth occurs forward at the coronal suture and backward at the lambdoid suture giving respectively a prominent forehead, called frontal bossing, and a prominent back portion of the head, called coning.[10][11] This is the most common form of craniosynostosis.[13]
Trigonocephaly
Trigonocephaly is a result from the premature closure of the metopic suture.[10][11] Using Virchow's law again to predict the resulting deformity, this fusion will result in a narrow forehead, which is even further emphasized by ridging of the suture.[10][11] Compensatory growth occurs at both the coronal sutures, thereby pushing the forehead forwards.[10][11] The resulting shape can best be assessed from a top view again, which will reveal a somewhat triangular form of the head.[12] Trigonocephaly is also a Greek-derived word, which can be translated as triangular-shaped head. A facial feature of metopic synostosis is hypotelorism; in the frontal view, it can be seen that the width between the eyes is smaller than usual.[11]
Plagiocephaly

The Greek word πλάγιος plágios means "skew". Plagiocephaly can be subclassified as anterior plagiocephaly or posterior plagiocephaly.
Anterior plagiocephaly
Anterior plagiocephaly is a clinical description of unilateral coronal synostosis.[10][11] Children born with unilateral coronal synostosis develop due to compensatory mechanisms a skew head; a plagiocephaly.[10][11]
The sagittal suture 'divides' the coronal suture in two halves; unilateral meaning that either the right side or the left side to the sagittal suture is fused. This fact immediately raises an important point. Unlike closure of the sagittal or the metopic suture, right and left are not the same in unilateral coronal synostosis.[10][11] This asymmetry shows in the skull deformity, as well as in the facial deformity and the complications.[10][11]
This time, the skull deformity can only partly be predicted using Virchow's law. Growth is arrested in the plane perpendicular to the fused suture and the forehead is flattened, but only at the ipsilateral side of the head.[10][11] Ipsilateral indicates the same side of the head as where the suture is closed. Compensatory growth occurs in a parallel plane, as well as in a perpendicular plane.[10][11] An increase in growth at the metopic and the sagittal suture accounts for the parallel plane and will result in bulging at the temporal fossa.[10][11] Compensatory growth in the perpendicular plane occurs on the side of the head with the patent coronal suture, the contralateral side.[10][11] Half of the forehead will bulge forwards.
Assessment of the skull from a top view shows asymmetry of the frontal bones, an increased width of the skull and a forward displacement of the ear at the ipsilateral side of the head.[12] Assessment of the skull from a frontal view will show asymmetrical features of the face, including a displacement of the chin point of the jaw and a deviation of the tip of the nose.[10][11] The chin point is located more to the contralateral side of the head, due to the ipsilateral forward displacement of the temporomandibular joint together with the ear.[10][11] The tip of the nose will also point towards the contralateral side.[10][11] Complications based on the skull deformation include malocclusion of the jaw, in as many as 90%; a subtle form of strabismus, caused by the asymmetrical placement of the orbits;[11] and refractive error, particularly astigmatism, due to asymmetrical development of the orbits.[14]
Posterior plagiocephaly
Unilateral lambdoid synostosis is also called posterior plagiocephaly, indicating that this gives, just like unilateral coronal synostosis, a 'skew head'. The difference is that this time, the deformity mostly shows at the occiput.
By Virchow's law, restriction of growth will occur at the ipsilateral side of the head; compensatory growth will occur at the contralateral side of the head. This growth pattern exerts an effect at the base of the skull, which is not even when the child is assessed from a point of view standing behind the child, as well as on the cervical spine, which shows a curvature.[15] In addition, from a point of view standing behind the child, a bulging of the mastoid can be seen.[15] Minimal forehead asymmetries are typically seen.[11]
Brachycephaly
Brachycephaly, or a 'short head', is the result of a closure of both the coronal sutures.[11] Following Virchow's law, this will result in a child's head with a restriction of growth in the forward direction and in the backward direction: recessed frontal bones and a flattened occiput.[11] Compensatory growth will occur sideways, due to the sagittal suture, and upwards, due to the lambdoid sutures.[11]
Oxycephaly
Oxycephaly, also known as turricephaly and high-head syndrome, is a type of cephalic disorder. This is a term sometimes used to describe the premature closure of the coronal suture plus any other suture, like the lambdoid suture,
Pansynostosis

The word pansynostosis is also Greek-derived and can be translated as "all one bone", indicating that all of the sutures are closed.[16] In general practice, the term is used to describe the children with three or more cranial sutures closed.[16]
Pansynostosis can present in several ways. The appearance can be the same as that seen with primary microcephaly: a markedly small head, but with normal proportions.[17] However, pansynostosis can also appear as a Kleeblattschädel (cloverleaf skull), which presents with bulging of the different bones of the cranial vault.[17]
The condition is associated with syndromes caused by mutations in fibroblast growth factor receptor genes (FGFR), including thanatophoric dwarfism type 2 (FGFR3) and Pfeiffer syndrome type 2 (FGFR2).
Other forms
- Apert syndrome: an abnormal skull shape, small upper jaw, and fusion of the fingers and toes.
- Crouzon syndrome: Craniofacial abnormalities with bilateral coronal suture fusion; anterior and posterior of skull shortness, flat cheek bones and a flat nose are their features.
- Muenke syndrome: Coronal craniosynostosis (plagiocephaly and brachycephaly), short feet and palms, hearing impairment, hypertelorism, and proptosis[18]
- Pfeiffer syndrome: abnormalities of the skull, hands, and feet; wide-set, bulging eyes, an underdeveloped upper jaw, beaked nose.
- Saethre–Chotzen syndrome: short or broad head; the eyes may be spaced wide apart and have palpebral ptosis (droopy eyelids), and fingers maybe abnormally short and webbed.[19][20]
 Apert syndrome
Apert syndrome.jpg.webp) Crouzon syndrome
Crouzon syndrome Muenke syndrome
Muenke syndrome Pfeiffer syndrome
Pfeiffer syndrome
Complications
Not all cranial abnormalities seen in children with craniosynostosis are solely a consequence of the premature fusion of a cranial suture. This is especially true in the cases with syndromic craniosynostosis. Findings include elevation of the intracranial pressure; obstructive sleep apnea (OSA); abnormalities in the skull base and neurobehavioral impairment.[11]
Elevated intracranial pressure
When the ICP is elevated the following symptomes may occur: vomiting, visual disturbance, bulging of the anterior fontanel, altered mental status, papilledema and headache.[21]
The main risks of prolonged elevated intracranial pressure may include cognitive impairment and impaired vision through prolonged papilledema[17] and subsequent optic atrophy.[22] These are the main reasons why fundoscopy should be performed during the physical examination of children with craniosynostosis.[17]
The causes of an elevation of the intracranial pressure are best understood using the Monro-Kellie doctrine.[23] The Monro-Kellie doctrine reduces the cranial vault to a box with rigid walls.[23] This box contains three elements: brain, intracranial blood and cerebrospinal fluid.[23] The sum of volumes of these three elements is constant.[23] An increase in one should cause a decrease in one or both of the remaining two, thereby preventing an elevation of the intracranial pressure.[23]
A compensatory mechanism involves the movement of cerebrospinal fluid from the cranial vault towards the spinal cord.[23] The volume of blood in the cranial vault is auto-regulated by the brain, and will therefore not decrease that easily.[23]
Intracranial pressure will rise as a result of continued brain growth within the rigid skull.[17] It appears that in children with craniosynostosis, the expected decrease of intracranial blood is probably not occurring as it should according to the Monro-Kellie hypothesis.[24] This is shown when the brain expands in the fixed skull, which gives a faster rise in intracranial pressure than would be expected.[24]
Obstructive sleep apnea
The short stops in breathing during the sleep are the mainstay of OSA. Other symptoms can be difficulty in breathing, snoring, day-time sleepiness and perspiration.[5] The main causative agent of OSA is the [midface hypoplasia], which also poses a risk to the eyes that can be seen bulging out of the eye sockets. Other factors, such as a micrognathism and adenoid hypertrophy, are likely to contribute in causing OSA.[5] The most common syndromic forms of craniosynostosis; i.e. Apert, Crouzon and Pfeiffer, have an increased risk of developing OSA. The children have nearly 50% chance of developing this condition.[5]
A theory regarding the involvement of OSA as a causative agent for elevated intracranial pressure suggests an association with the auto-regulation of blood flow in the brain.[25] Certain cells in the brain respond specifically to an increase of CO2 in the blood.[4][26] The response involves vasodilatation of the cranial vault blood vessels, increasing the volume of one of the elements in the Monro-Kellie doctrine.[4][26] The increase of CO2 concentration in the blood is a consequence of impaired breathing, especially seen when the child with OSA is sleeping.[4][26] It is well documented that the highest spikes in intracranial pressure often occur during sleep.[4][26]
Abnormalities in the skull base
Impaired venous outflow is often caused by a hypoplastic jugular foramen.[25] This causes an increase in the intracranial blood volume, thereby causing an increase in intracranial pressure.[25]
This can be further complicated with a possible Arnold–Chiari malformation, which can partially obstruct the flow of cerebro-spinal fluid from the neurocranium to the spinal cord.[6] The Chiari malformation may be asymptomatic or present with ataxia, spasticity or abnormalities in breathing, swallowing or sleeping.[6]
Due to the impaired venous outflow, which may be further complicated with an Arnold–Chiari malformation, there is often a clinical image of hydrocephalus present. Hydrocephalus is seen in 6.5 to 8% of patients with Apert's syndrome, 25.6% in patients with Crouzon's syndrome and 27.8% of those with Pfeifer's syndrome.[27] Ventriculomegaly is a usual finding in children with the Apert syndrome.[28]
Neurobehavioural impairment
Neurobehavioural impairment includes problems with attention and planning, processing speed, visual spatial skills, language, reading and spelling.[10] A decreased IQ may also be part of the problems.[10]
It has been suggested that these problems are caused by a primary malformation of the brain, rather than being a consequence of the growth restriction of the skull and elevated intracranial pressure. Some evidence for this statement has been provided by studies using computed tomographic (CT) scans and magnetic resonance imaging (MRI) to identify differences between the structures of the brains of healthy children and those affected with craniosynostosis.[10] It has been found that corrective surgery of the cranial vault alters the morphology of the brain compared with the situation before surgical intervention.[10] However the structure was still abnormal in comparison to children without craniosynostosis.[10]
Causes
Advances in the fields of molecular biology and genetics, as well as the use of animal models have been of great importance in expanding our knowledge of suture fusion.[3] Research in animal models has led to the idea that the dura mater plays an important role in determining closure or patency of the suture.[3] In contrast to the dura mater it appears that the periosteum is not essential in causing closure or patency.[3]

Instead of describing the abnormalities in structure and form, research focuses nowadays at decoding the molecular interactions that underlie them.[3] Based on data from quantitative real-time PCR on samples of suture junctions during development, cranial suture fusion in mammals is a tightly orchestrated expression of genes in specific temporal order, leading to endochondral ossification.[29] Despite the progress that has been made, many things are still not understood about the suture biology and the exact causative pathways remain yet to be completely understood.[30]
Multiple potential causes of premature suture closure have been identified, such as the several genetic mutations that are associated with syndromic craniosynostosis.[3] The cause of nonsyndromic craniosynostosis however, is still greatly unknown.[10] Most likely, a role is played by biomechanical factors, as well as environmental, hormonal and genetical factors.[10]
New insights have given fuel to a debate whether there might be an intrinsic factor causing the premature fusion of the sutures. Brain structures of children with craniosynostosis were evaluated using magnetic resonance imaging.[10] Differences were seen compared with the brain structures of normal children.[10] The question now is whether these differences are caused by the craniosynostosis, or are the cause of craniosynostosis.
Biomechanical factors
Biomechanical factors include fetal head constraint during pregnancy.[31] It has been found by Jacob et al. that constraint inside the womb is associated with decreased expression of Indian hedgehog protein and noggin. These last two are both important factors influencing bone development.[31]
Environmental factors
Environmental factors refer for example to maternal smoking and the maternal exposure to amine-containing drugs. Several research groups have found evidence that these environmental factors are responsible for an increase in the risk of craniosynostosis, likely through effects on fibroblast growth factor receptor genes.[32][33][34][35][36]
On the other hand, a recent evaluation of valproic acid (an anti-epilepticum), which has been implicated as a causative agent, has shown no association with craniosynostosis.[37]
Certain medication (like amine-containing drugs) can increase the risk of craniosynostosis when taken during pregnancy, these are so-called teratogenic factors.[33][36]
Hormonal factors
Hyperthyroid induced craniosynostosis is a hormone mediated premature closure.[38] It is thought that the bone matures faster due to high levels of thyroid hormone.[38]
Genetic factors
In 6 to 11% of the children born with coronal synostosis, more often involving the bilateral cases than unilateral, other members of the family have been reported that were also born with the same condition.[39] This finding is highly suggestive of a genetic cause, which has possibly been found in the fibroblast growth factor receptor 3 (FGFR3) and TWIST genes.[39]
Fibroblast growth factor and fibroblast growth factor receptors regulate fetal bone growth and are expressed in cranial sutures during pregnancy.[6] The transcription factor gene TWIST is thought to decrease the function of FGFR, thus also indirectly regulating fetal bone growth.[6] A relation between the mutations in these genes and craniosynostosis is therefore possible. Moloney et al. observed a FGFR3 mutation in as many as 31% of the cases with nonsyndromic coronal synostosis, thus showing that FGFR abnormalities play an important role in nonsyndromic craniosynostosis.[40]
In terms of syndromic craniosynostosis not only do FGFR3 and TWIST genes feature, but also FGFR1 and in particular FGFR2, which has been reported in 90% of the syndromic craniosynostoses such as Apert, Crouzon, Peiffer and Jackson–Weiss.[41][42][43] The mutations can be divided into mutations that lead to gain of function (in FGFR genes) and mutations that lead to loss of function (in TWIST genes).[42][43] Craniosynostosis is therefore likely the result of a disturbance in the fine balance that regulates the multiplication and maturation of the precursor bone cells in the cranial sutures.[3]
The familial rate, which is different for nonsyndromic and syndromic cases, provides an important clue.[44][45] In the nonsyndromic cases, a positive family history is found in 2% of the cases with sagittal suture closure[44][45] and in 6% to 11% of the cases with coronal suture closure.
Cranial sutures

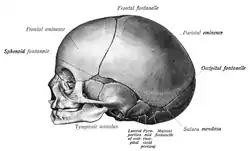
The mesenchyme above the meninges undergoes intramembranous ossification forming the neurocranium.[3] The neurocranium consists of several bones, which are united and at the same time separated by fibrous sutures.[3] This allows movement of the separate bones in relation to one another; the infant skull is still malleable.[3] The fibrous sutures specifically allow the deformation of the skull during birth[3] and absorb mechanical forces during childhood[6] They also allow the necessary expansion during brain growth.[3]
In the very first years of life the sutures serve as the most important centers of growth in the skull.[3] The growth of the brain and the patency of the sutures depend on each other.[46] Brain growth pushes the two sides of the patent sutures away from each other, thereby enabling growth of the neurocranium.[46] This means that the neurocranium can only grow if the sutures remain open.[46] The neurocranium will not grow when the forces induced by brain growth are not there.[12] This will occur for example when the intracranial pressure drops; the sutures do not experience stretching anymore causing them to fuse.[17]
Diagnosis
The evaluation of a child suspected to have craniosynostosis is preferentially performed in a craniofacial center. The three main elements of analysis include medical history, physical examination and radiographic analysis.[47]
Medical history should in any case include questions about risk factors during pregnancy, the familial rate and the presence of symptoms of elevated intracranial pressure (ICP).
Elevated ICP
- Symptoms of increased intracranial pressure – such as headache and vomiting – should be questioned after.[24][25] An elevation of ICP can be present in 4 to 20% of the children where only a single suture is affected.[24][25] The incidence of ICP in children with more than one suture involved can be as high as 62%.[48] However, even though the children are affected, symptoms are not always present.
Physical examination
Fundoscopy should always be performed in children with craniosynostosis.[49] It is used to find papilledema which is sometimes the only symptom of elevated intracranial pressure shown in these children.[49]
Other parts of the physical examination include the measurement of the head circumference, the assessment of the skull deformity and the search for deformities affecting other parts of the body.[17] The head circumference and the growth curve of the head provide important clues into making a differentiation between craniosynostosis, primary microcephaly and hydrocephalus.[17] This differentiation has an important influence on the further treatment of the child.[17]
In a recent article Cunningham et al.[12] described several steps in which a pediatrician should observe the patient to assess skull deformity:
- The first is looking with a bird's eye view at the patient while the patient preferably faces the parent while sitting on the parent's lap. The goal is to assess the shape of the forehead, the skull length, the width of the skull, position of the ears and the symmetry of the frontal bones and [occiput].
- The second is looking at the patient from behind while preferably the child is in the same position as described above. It is important to look at the skull base (to determine whether it is level or not), the position of the ears and to the mastoid (to spot the possible presence of a bulge).
- The third point of view is the frontal view. The points to look at are: eye position, eye symmetry and twisting of the nasal tip.
The implications of the deformities that are seen are extensively discussed under 'phenotype'.
Syndromal craniosynostosis presents with a skull deformity as well as deformities affecting other parts of the body.[46] Clinical examination should in any case include evaluation of the neck, spine, digits and toes.[46]
Medical imaging
Radiographic analysis by performing a computed axial tomographic scan is the gold standard for diagnosing craniosynostosis.[50][51]
Plain radiography of the skull may be sufficient for diagnosing a single suture craniosynostosis and should therefore be performed,[50][51] but the diagnostic value is outweighed by that of the CT-scan.[52] Not only can the sutures be identified more accurately, thus objectively demonstrating a fused suture, but also evaluation of the brain for structural abnormalities and excluding other causes of asymmetric growth are possible at the same time.[52] In addition to this, CT-scanning can visualize the extent of skull deformity, thereby enabling the surgeon to start planning surgical reconstruction.[53]
Classification
There are several ways to classify craniosynostosis.
- For example, one can consider the number of closed sutures. If only one of the four sutures is prematurely closed (single suture craniosynostosis), the craniosynostosis is referred to as 'simple' (or 'isolated'). Whereas when two or more sutures are no longer open, the craniosynostosis is 'complex'.[6]
- A second classification scheme gives a clinical description of the resulting shape of the skull. This will be further discussed under phenotype.
- A third classification involves the presence or absence of an identified craniofacial syndrome. Craniosynostosis where no extracranial deformations are present, is called non-syndromic or 'isolated' craniosynostosis.[6] When there are extracranial deformations present, for instance involving the limbs, heart, central nervous system or the respiratory tract,[12] you may speak of a syndromic form of craniosynostosis. More than 180 identified syndromes show deformations due to craniosynostosis.[6] The following syndromes are associated with fibroblast growth factor receptors:
| Name of syndrome | Other signs and symptoms (along with craniosynostosis; may not all be present) | OMIM reference | Gene |
|---|---|---|---|
| Crouzon syndrome | wide-set, bulging eyes • beaked nose • flat face | 123500 | FGFR2, FGFR3 |
| Apert syndrome | fused fingers or toes • flat midface | 101200 | FGFR2 |
| Crouzonodermoskeletal syndrome | wide-set, bulging eyes • beaked nose • flat face • dark, velvety skin folds • spine abnormalities • benign growths in the jaw | 134934 | FGFR3 |
| Jackson–Weiss syndrome | enlarged, bent big toes • flat midface | 123150 | FGFR1, FGFR2 |
| Muenke syndrome | coronal synostosis • skeletal abnormalities of the hands or feet • hearing loss | 602849 | FGFR3 |
| Pfeiffer syndrome | broad, short thumbs or big toes • webbed or fused fingers or toes | 101600 | FGFR1, FGFR2 |
In addition, the following syndromes have been identified:
| Name of syndrome | Other signs and symptoms (along with craniosynostosis; may not all be present) | OMIM reference | Gene |
|---|---|---|---|
| Loeys–Dietz syndrome | wide-set eyes • split uvula or cleft palate • arterial tortuosity • aortic root dilatation • aneurysms | 609192 610168 613795 608967 610380 | TGFBR1, TGFBR2, SMAD3 |
| Saethre–Chotzen syndrome | facial asymmetry • low frontal hairline • drooping eyelids • webbed fingers or toes • broad big toes | 101400 | TWIST1 |
| Shprintzen–Goldberg syndrome | bulging eyes • flat face • hernias • long, thin fingers • developmental delay • intellectual disability | 182212 | FBN1 |
Deformational plagiocephaly
The main difference between plagiocephaly based on craniosynostosis and deformational plagiocephaly is that there is no suture fusion in the latter one.[12] The malleability of the neonatal skull allows the skull to change shape due to extrinsic forces.[12]
With the tests a pediatrician should perform, as explained above, the difference is quite easy to make.[12] In deformational plagiocephaly the skull does not show a bulging of the mastoid and in these patients the skull base and position of the ears is level, all in contrary with plagiocephaly due to craniosynostosis.[12] Displacement of one ear to the front is characteristic for deformational plagiocephaly.[12]
Primary microcephaly
Primary microcephaly shows a clinical image of craniosynostosis, but due to a different cause. The primary failure is the absence of growth of the brain, rendering the sutures of the cranial vault useless.[17] As a consequence, the sutures close, presenting a pansynostosis like image.[17] A differentiation between these two conditions can be made with a computed tomography (CT) scan. The subarachnoid spaces are typically enlarged with primary microcephaly, whereas they are reduced or absent in true pansynostosis.[17]
Treatment
The primary goal of surgical intervention is to allow normal cranial vault development to occur.[46] This can be achieved by excision of the prematurely fused suture and correction of the associated skull deformities.[46] If the synostosis goes uncorrected, the deformity will progressively worsen not only threatening the aesthetic aspect, but also the functional aspect.[46] This is especially prevalent with asymmetric conditions, such as unilateral coronal synostosis, with compromised function of the eyes and the jaw.[46] In addition, signs of compromised neurodevelopment have been seen amongst all the synostoses, although this may also be caused by primary maldevelopment of the brain and can thus not be prevented by surgical intervention.[54]
There are a few basic elements involved in surgical intervention which is aimed at normalization of the cranial vault:
- Surgery for craniosynostosis is often associated with significant perioperative hemorrhage so multiple strategies are often used to minimize blood loss.[55] One such method involves the injection of vasoconstrictive agents (i.e. epinephrine) seven to ten minutes before scalp incision.[46] Additionally, the initiation of surgery should be delayed until blood products are physically present in the operating room.[46]
- Another general agreement is the avoidance of the use of titanium plates in the fixation of the skull.[56][57] One potential complication following this procedure involves the gradual movement of the titanium plates towards the brain, induced by resorption of the innermost bone layer of the skull with deposition of new bone on the outermost layer, thereby integrating the titanium plates.[56][57] In some cases, the plates have been observed to come into direct contact with the brain.[56][57] Absorbable plates are now used instead.[56][57]
Timing of surgery
The prevention of post-surgical complications, including those mentioned above, plays an important role in discussions over the timing of potential surgery. The general consensus is to perform surgery in late infancy, i.e. between six and twelve months.[46] Within this time frame the efficacy of surgery will be enhanced for several reasons:
- The bone is still more malleable and can be remodelled relatively 'simply' by greenstick fractures of the bone.[46] At approximately one year of age the bone has become more mineralized and brittle and needs to be attached to the surrounding bone with sutures or an absorbable plate.[46]
- Reshaping of the cranial vault most commonly means excision of the bones to allow shape adjustment.[46] Replacement of cranial bones can leave 'gaps' which are readily re-ossified before the age of one year, but will need bony filling thereafter.[46]
Most surgeons will not intervene until after the age of six months due to the heightened risk which blood loss poses before this age.[46] It is generally preferable to wait until after three months of age when anaesthetic risks will also be decreased.[46]
Surgery is not performed in early childhood in every country; in some countries surgical intervention can take place in the late teens.
It is important that families seek out a Pediatric Craniofacial Physician who has experience with craniosynostosis for proper diagnosis, surgical care, and followup.
Sagittal craniosynostosis/scaphocephaly
There are two surgical procedures which are commonly used to treat sagittal synostosis.[46] The matter of which procedure is superior is still heavily debated amongst the surgeons treating this condition,[46] however it is generally agreed upon that the cephalic index should be used to assess the efficacy of the preferred surgical intervention.[46]
- Firstly, the extended strip craniectomy will be discussed, which is a further developed form of the traditional craniectomy.[46] The traditional procedure involved only the excision of the closed suture, with the intent that the compensatory skull deformations would automatically be corrected by the fast growth of the brain during the first year of life.[46]
This did not quite result in a true normalization of the cranial vault, causing the development of numerous modifications.[58][59] One of those is the extended strip craniectomy. This procedure can be performed by using an endoscope, becoming more popular due to the resultant rapid recovery of the child and reduced need for blood transfusion.[58][59] The patient is afterwards placed in a custom made molding helmet to correct the resting deformities of the skull.[58][59] - In the other widely used surgical procedure, total cranial vault remodelling, the compensatory deformities are corrected during the operation, with excision of the fused suture.[60] Most of the bones that collectively form the cranial vault – i.e. the frontal, the parietal and the occipital bones – are removed and reshaped to normalize the contours.[60] The bones are then replaced and fixated to form a cranial vault with a normalized shape.[60]
Retrospective analysis has given an indication that the use of total cranial vault remodelling provides children with a better cephalic index than does the extended strip craniectomy.[61]
An approach that is currently being evaluated involves the use of springs. This intervention is likely most effective when used in the time frame between three and six months of age.
Metopic synostosis/trigonocephaly
The main elements of metopic suture closure involve a low volume of the anterior cranial fossa, the metopic ridging and hypotelorism.[62] These problems are all addressed during the surgical intervention.[62]
The volume is increased by placing the frontal bones and the supraorbital rim further forward.[46] This is done by excision of the bones after which they are reshaped with greenstick fracturing.[46] Replacement of the bones provides a possibility for the correction of the hypotelorism at the same time.[46] A bone graft is placed in between the two halves of the supraorbital bars, thereby increasing the width between the orbits.[46] The metopic ridge can then be corrected with a (simple) burring.[11]
Unilateral coronal synostosis/anterior plagiocephaly
The treatment of unilateral coronal synostosis is typically performed in two parts: the forward advancement of the supraorbital bar and the correction of the orbital asymmetry.[46]
The supraorbital bar is the rim just above the eye socket; as discussed under phenotype, the supraorbital and the frontal bone are typically recessed at the ipsilateral side of the head.[46] The goal of treatment is to position this bar together with the frontal bone in a plane three millimetres further forwards than the vertical plane of the cornea.[46] A two-dimensional sagittal image is used to pre-operatively determine the extent of movement, which can vary between seven and fifteen millimetres depending on the severity of the deformity.[46]
The orbital asymmetry exists typically of a narrower and taller orbit at the ipsilateral side of the head.[46] The contralateral orbit, however, is wider than usual.[46] Symmetry is restored by extracting a small piece of bone from the supraorbital bar at the contralateral side, thereby reducing the width.[46] This bone fragment is then introduced into the supraorbital bar on the ipsilateral side, thereby increasing width.[46] The height of the orbit is altered at the ipsilateral side only, by extracting a piece of bone.[46] Any correction of the nasal tip, which points towards the contralateral side, will not be performed during childhood.[11]
Unilateral lambdoid synostosis/posterior plagiocephaly
An excision of the flattened occipital bone with release of the fused suture tends to correct the cranial vault deformity.[11]
Bilateral coronal synostosis/brachycephaly
The treatment of bilateral coronal synostosis shows a high degree of overlap with treatment of unilateral coronal synostosis; in both surgical interventions is the forward advancement of the supraorbital rim together with the frontal bones is an important part of the procedure.[11][46] Again, the plane three millimetres further forwards than the vertical plane of the [cornea] is the appropriate position to place the bone.[11][46] The increased height of the skull is addressed in the same procedure.[11] Parts of the flattened occiput are extracted and given a rounder shape by greenstick fracturing them.[11]
Pansynostosis/kleeblattschädel
The treatment of pansynostosis comprises the expansion of the anterior cranial vault, as well as the posterior cranial vault.[17] Whilst this may be accomplished in one procedure it is generally performed in two stages.[17]
Complications
The most common complications of uncorrected craniosynostosis include increased intracranial pressure, asymmetry of the face, and malocclusion. Asymmetry of the orbits often leads to strabismus.[63]
Epidemiology
It is estimated that craniosynostosis affects 1 in 1,800 to 3,000 live births worldwide.[3] three out of every four cases affect males. Sagittal synostosis is the most common phenotype, representing 40% to 55% of nonsyndromic cases,[3] whilst coronal synostosis represents between 20% to 25% of cases.[3] Metopic synostosis is a factor in 5% to 15% of cases, and lambdoid synostosis is seen in 0% to 5% of nonsyndromic cases.[3]
Five to 15% of the time more than one suture is involved; this is referred to as "complex craniosynostosis" and is typically part of a syndrome.[3]
References
- Khanna PC, Thapa MM, Iyer RS, Prasad SS (January 2011). "Pictorial essay: The many faces of craniosynostosis". The Indian Journal of Radiology & Imaging. 21 (1): 49–56. doi:10.4103/0971-3026.76055. PMC 3056371. PMID 21431034.
- Silva S, Jeanty P (1999-06-07). "Cloverleaf skull or kleeblattschadel". TheFetus.net. MacroMedia. Archived from the original on 2007-01-07. Retrieved 2007-02-03.
- Slater BJ, Lenton KA, Kwan MD, Gupta DM, Wan DC, Longaker MT (April 2008). "Cranial sutures: a brief review". Plastic and Reconstructive Surgery. 121 (4): 170e–8e. doi:10.1097/01.prs.0000304441.99483.97. PMID 18349596. S2CID 34344899.
- Gault DT, Renier D, Marchac D, Jones BM (September 1992). "Intracranial pressure and intracranial volume in children with craniosynostosis". Plastic and Reconstructive Surgery. 90 (3): 377–81. doi:10.1097/00006534-199209000-00003. PMID 1513883.
- Bannink N, Nout E, Wolvius EB, Hoeve HL, Joosten KF, Mathijssen IM (February 2010). "Obstructive sleep apnea in children with syndromic craniosynostosis: long-term respiratory outcome of midface advancement". International Journal of Oral and Maxillofacial Surgery. 39 (2): 115–21. doi:10.1016/j.ijom.2009.11.021. PMID 20056390.
- Kimonis V, Gold JA, Hoffman TL, Panchal J, Boyadjiev SA (September 2007). "Genetics of craniosynostosis". Seminars in Pediatric Neurology. 14 (3): 150–61. doi:10.1016/j.spen.2007.08.008. PMID 17980312.
- Kabbani H, Raghuveer TS (June 2004). "Craniosynostosis". American Family Physician. 69 (12): 2863–70. PMID 15222651.
- Delashaw JB, Persing JA, Broaddus WC, Jane JA (February 1989). "Cranial vault growth in craniosynostosis". Journal of Neurosurgery. 70 (2): 159–65. doi:10.3171/jns.1989.70.2.0159. PMID 2913214.
- Agrawal D, Steinbok P, Cochrane DD (March 2005). "Scaphocephaly or dolichocephaly?". Journal of Neurosurgery. 102 (2 Suppl): 253–4, author reply 254. doi:10.3171/jns.2005.102.2.0253. PMID 16156241.
- Kapp-Simon KA, Speltz ML, Cunningham ML, Patel PK, Tomita T (March 2007). "Neurodevelopment of children with single suture craniosynostosis: a review". Child's Nervous System. 23 (3): 269–81. doi:10.1007/s00381-006-0251-z. PMID 17186250. S2CID 29722887.
- Persing JA (April 2008). "MOC-PS(SM) CME article: management considerations in the treatment of craniosynostosis". Plastic and Reconstructive Surgery. 121 (4 Suppl): 1–11. doi:10.1097/01.prs.0000305929.40363.bf. PMID 18379381.
- Cunningham ML, Heike CL (December 2007). "Evaluation of the infant with an abnormal skull shape". Current Opinion in Pediatrics. 19 (6): 645–51. doi:10.1097/MOP.0b013e3282f1581a. PMID 18025930. S2CID 26966396.
- "Sagittal Craniosynostosis". www.stlouischildrens.org. Retrieved 2022-04-16.
- Levy, Richard Lawrence; Rogers, Gary F.; Mulliken, John B.; Proctor, Mark R.; Dagi, Linda R. (2007). "Astigmatism in unilateral coronal synostosis: Incidence and laterality". J AAPOS. 11 (4): 367–372. doi:10.1016/j.jaapos.2007.02.017. PMID 17588790.
- Huang MH, Gruss JS, Clarren SK, Mouradian WE, Cunningham ML, Roberts TS, Loeser JD, Cornell CJ (October 1996). "The differential diagnosis of posterior plagiocephaly: true lambdoid synostosis versus positional molding". Plastic and Reconstructive Surgery. 98 (5): 765–74, discussion 775–6. doi:10.1097/00006534-199610000-00002. PMID 8823012.
- Chumas PD, Cinalli G, Arnaud E, Marchac D, Renier D (February 1997). "Classification of previously unclassified cases of craniosynostosis". Journal of Neurosurgery. 86 (2): 177–81. doi:10.3171/jns.1997.86.2.0177. PMID 9010415.
- Blount JP, Louis RG, Tubbs RS, Grant JH (October 2007). "Pansynostosis: a review". Child's Nervous System. 23 (10): 1103–9. doi:10.1007/s00381-007-0362-1. PMID 17486351. S2CID 43068552.
- "Muenke Syndrome - About the Disease - Genetic and Rare Diseases Information Center". rarediseases.info.nih.gov. Retrieved 2022-07-10.
- "Types of Craniosynostosis". Craniosynostosis Surgery in South Texas.
- "What is Craniosynostosis?". The Johns Hopkins Hospital, and Johns Hopkins Health System.
- de Jong T, Bannink N, Bredero-Boelhouwer HH, van Veelen ML, Bartels MC, Hoeve LJ, Hoogeboom AJ, Wolvius EB, Lequin MH, van der Meulen JJ, van Adrichem LN, Vaandrager JM, Ongkosuwito EM, Joosten KF, Mathijssen IM (October 2010). "Long-term functional outcome in 167 patients with syndromic craniosynostosis; defining a syndrome-specific risk profile". Journal of Plastic, Reconstructive & Aesthetic Surgery. 63 (10): 1635–41. doi:10.1016/j.bjps.2009.10.029. PMID 19913472.
- Newman, Steven A. (1991). "Ophthalmic Features of Cransosynostosis". Neurosurgery Clinics of North America. 2 (3): 587–610. doi:10.1016/S1042-3680(18)30721-6. PMID 1821306.
- Mokri B (June 2001). "The Monro-Kellie hypothesis: applications in CSF volume depletion". Neurology. 56 (12): 1746–8. doi:10.1212/WNL.56.12.1746. PMID 11425944. S2CID 1443175.
- Tamburrini G, Caldarelli M, Massimi L, Santini P, Di Rocco C (October 2005). "Intracranial pressure monitoring in children with single suture and complex craniosynostosis: a review". Child's Nervous System. 21 (10): 913–21. doi:10.1007/s00381-004-1117-x. PMID 15871027. S2CID 6442539.
- Bristol RE, Lekovic GP, Rekate HL (December 2004). "The effects of craniosynostosis on the brain with respect to intracranial pressure". Seminars in Pediatric Neurology. 11 (4): 262–7. doi:10.1016/j.spen.2004.11.001. PMID 15828710.
- Eide PK (2006). "Assessment of quality of continuous intracranial pressure recordings in children". Pediatric Neurosurgery. 42 (1): 28–34. doi:10.1159/000089506. PMID 16357498. S2CID 32754729.
- Cohen MM, MacLean RE (2000). Craniosynostosis: Diagnosis, Evaluation, and Management (2nd ed.). Oxford: Oxford University Press. pp. 316–353. ISBN 978-0-19-511843-8.
- Collmann H, Sörensen N, Krauss J (October 2005). "Hydrocephalus in craniosynostosis: a review". Child's Nervous System. 21 (10): 902–12. doi:10.1007/s00381-004-1116-y. PMID 15864600. S2CID 9767488.
- Sahar DE, Longaker MT, Quarto N. Sox9 neural crest determinant gene controls patterning and closure of the posterior frontal cranial suture. Dev Biol. 2005 Apr 15;280(2):344-61.
- Choi, M.D., Jung Won (May 10, 2016). "Craniosynostosis in Growing Children : Pathophysiological Changes and Neurosurgical Problems". Journal of Korean Neurosurgical Society. 59 (3): 197–203. doi:10.3340/jkns.2016.59.3.197. PMC 4877540. PMID 27226849.
- Jacob S, Wu C, Freeman TA, Koyama E, Kirschner RE (February 2007). "Expression of Indian Hedgehog, BMP-4 and Noggin in craniosynostosis induced by fetal constraint". Annals of Plastic Surgery. 58 (2): 215–21. doi:10.1097/01.sap.0000232833.41739.a5. PMID 17245153. S2CID 43913649.
- Carmichael SL, Ma C, Rasmussen SA, Honein MA, Lammer EJ, Shaw GM (February 2008). "Craniosynostosis and maternal smoking". Birth Defects Research. Part A, Clinical and Molecular Teratology. 82 (2): 78–85. doi:10.1002/bdra.20426. PMID 18050313.
- Gardner JS, Guyard-Boileau B, Alderman BW, Fernbach SK, Greene C, Mangione EJ (February 1998). "Maternal exposure to prescription and non-prescription pharmaceuticals or drugs of abuse and risk of craniosynostosis". International Journal of Epidemiology. 27 (1): 64–7. doi:10.1093/ije/27.1.64. PMID 9563695.
- Honein MA, Rasmussen SA (September 2000). "Further evidence for an association between maternal smoking and craniosynostosis". Teratology. 62 (3): 145–6. doi:10.1002/1096-9926(200009)62:3<145::AID-TERA1>3.0.CO;2-7. PMID 10935977.
- Källén K (September 1999). "Maternal smoking and craniosynostosis". Teratology. 60 (3): 146–50. doi:10.1002/(SICI)1096-9926(199909)60:3<146::AID-TERA10>3.0.CO;2-3. PMID 10471899.
- Olshan AF, Faustman EM (December 1989). "Nitrosatable drug exposure during pregnancy and adverse pregnancy outcome". International Journal of Epidemiology. 18 (4): 891–9. doi:10.1093/ije/18.4.891. PMID 2621027.
- Jentink J, Loane MA, Dolk H, Barisic I, Garne E, Morris JK, de Jong-van den Berg LT (June 2010). "Valproic acid monotherapy in pregnancy and major congenital malformations". The New England Journal of Medicine. 362 (23): 2185–93. doi:10.1056/NEJMoa0907328. PMID 20558369.
- Johnsonbaugh RE, Bryan RN, Hierlwimmer R, Georges LP (August 1978). "Premature craniosynostosis: A common complication of juvenile thyrotoxicosis". The Journal of Pediatrics. 93 (2): 188–91. doi:10.1016/S0022-3476(78)80493-4. PMID 209162.
- Mulliken JB, Gripp KW, Stolle CA, Steinberger D, Müller U (June 2004). "Molecular analysis of patients with synostotic frontal plagiocephaly (unilateral coronal synostosis)". Plastic and Reconstructive Surgery. 113 (7): 1899–909. doi:10.1097/01.prs.0000122202.26792.bf. PMID 15253176. S2CID 6382480.
- Moloney DM, Wall SA, Ashworth GJ, Oldridge M, Glass IA, Francomano CA, Muenke M, Wilkie AO (April 1997). "Prevalence of Pro250Arg mutation of fibroblast growth factor receptor 3 in coronal craniosynostosis". Lancet. 349 (9058): 1059–62. doi:10.1016/S0140-6736(96)09082-4. PMID 9107244. S2CID 38612577.
- Passos-Bueno MR, Serti Eacute AE, Jehee FS, Fanganiello R, Yeh E (2008). "Genetics of craniosynostosis: genes, syndromes, mutations and genotype–phenotype correlations". Frontiers of Oral Biology. 12 (1): 107–43. doi:10.1159/000115035. ISBN 978-3-8055-8326-8. PMID 18391498.
- Lenton KA, Nacamuli RP, Wan DC, Helms JA, Longaker MT (2005). "Cranial suture biology". Current Topics in Developmental Biology. 66: 287–328. doi:10.1016/S0070-2153(05)66009-7. ISBN 9780121531669. PMID 15797457.
- Wilkie AO (1997). "Craniosynostosis: genes and mechanisms". Human Molecular Genetics. 6 (10): 1647–56. doi:10.1093/hmg/6.10.1647. PMID 9300656.
- Hunter AG, Rudd NL (October 1976). "Craniosynostosis. I. Sagittal synostosis: its genetics and associated clinical findings in 214 patients who lacked involvement of the coronal suture(s)". Teratology. 14 (2): 185–93. doi:10.1002/tera.1420140209. PMID 982314.
- Lajeunie E, Le Merrer M, Bonaïti-Pellie C, Marchac D, Renier D (March 1996). "Genetic study of scaphocephaly". American Journal of Medical Genetics. 62 (3): 282–5. doi:10.1002/(SICI)1096-8628(19960329)62:3<282::AID-AJMG15>3.0.CO;2-G. PMID 8882788.
- Panchal J, Uttchin V (May 2003). "Management of craniosynostosis". Plastic and Reconstructive Surgery. 111 (6): 2032–48, quiz 2049. doi:10.1097/01.PRS.0000056839.94034.47. PMID 12711969.
- "Craniosynostosis | Boston Children's Hospital". www.childrenshospital.org. Retrieved 2022-07-25.
- Renier D, Lajeunie E, Arnaud E, Marchac D (November 2000). "Management of craniosynostoses". Child's Nervous System. 16 (10–11): 645–58. doi:10.1007/s003810000320. PMID 11151714. S2CID 22876385.
- Bannink N, Joosten KF, van Veelen ML, Bartels MC, Tasker RC, van Adrichem LN, van der Meulen JJ, Vaandrager JM, de Jong TH, Mathijssen IM (January 2008). "Papilledema in patients with Apert, Crouzon, and Pfeiffer syndrome: prevalence, efficacy of treatment, and risk factors". The Journal of Craniofacial Surgery. 19 (1): 121–7. doi:10.1097/SCS.0b013e31815f4015. PMID 18216676. S2CID 41004047.
- Cerovac S, Neil-Dwyer JG, Rich P, Jones BM, Hayward RD (August 2002). "Are routine preoperative CT scans necessary in the management of single suture craniosynostosis?". British Journal of Neurosurgery. 16 (4): 348–54. doi:10.1080/0268869021000007560. PMID 12389887. S2CID 23376632.
- Goldstein SJ, Kidd RC (1982). "Value of computed tomography in the evaluation of craniosynostosis". Computerized Radiology. 6 (6): 331–6. doi:10.1016/0730-4862(82)90003-8. PMID 7166030.
- Aviv RI, Rodger E, Hall CM (February 2002). "Craniosynostosis". Clinical Radiology. 57 (2): 93–102. doi:10.1053/crad.2001.0836. PMID 11977940.
- Ghali GE, Sinn DP, Tantipasawasin S (March 2002). "Management of nonsyndromic craniosynostosis". Atlas of the Oral and Maxillofacial Surgery Clinics of North America. 10 (1): 1–41. doi:10.1016/s1061-3315(01)00003-8. PMID 12087862.
- Speltz ML, Kapp-Simon KA, Cunningham M, Marsh J, Dawson G (December 2004). "Single-suture craniosynostosis: a review of neurobehavioral research and theory". Journal of Pediatric Psychology. 29 (8): 651–68. doi:10.1093/jpepsy/jsh068. PMID 15491988.
- White N, Bayliss S, Moore D (January 2015). "Systematic review of interventions for minimizing perioperative blood transfusion for surgery for craniosynostosis". The Journal of Craniofacial Surgery. 26 (1): 26–36. doi:10.1097/scs.0000000000001108. PMID 25569385. S2CID 37484366.
- Fearon JA, Munro IR, Bruce DA (April 1995). "Observations on the use of rigid fixation for craniofacial deformities in infants and young children". Plastic and Reconstructive Surgery. 95 (4): 634–7, discussion 638. doi:10.1097/00006534-199504000-00002. PMID 7892306. S2CID 23805140.
- Duke BJ, Mouchantat RA, Ketch LL, Winston KR (July 1996). "Transcranial migration of microfixation plates and screws. Case report". Pediatric Neurosurgery. 25 (1): 31–4, discussion 35. doi:10.1159/000121093. PMID 9055332.
- Jimenez DF, Barone CM (January 1998). "Endoscopic craniectomy for early surgical correction of sagittal craniosynostosis". Journal of Neurosurgery. 88 (1): 77–81. doi:10.3171/jns.1998.88.1.0077. PMID 9420076.
- Barone CM, Jimenez DF (December 1999). "Endoscopic craniectomy for early correction of craniosynostosis". Plastic and Reconstructive Surgery. 104 (7): 1965–73, discussion 1974–5. doi:10.1097/00006534-199912000-00003. PMID 11149758.
- Francel PC (1995). "Evolution of the treatment for sagittal synostosis: a personal record". In Goodrich JT, Hall CD (eds.). Craniofacial Anomalies: Growth and Development from a Surgical Perspective. New York: Thieme. ISBN 978-0-86577-522-0. OCLC 490069409.
- Panchal J, Marsh JL, Park TS, Kaufman B, Pilgram T, Huang SH (May 1999). "Sagittal craniosynostosis outcome assessment for two methods and timings of intervention". Plastic and Reconstructive Surgery. 103 (6): 1574–84. doi:10.1097/00006534-199905000-00004. PMID 10323690.
- Francel PC (1999). "Metopic synostosis". In Zumdahl SS (ed.). Neurosurgical Operative Atlas. Vol. 9. Rolling Meadows IL: American Association of Neurological Surgeons. pp. 91–111. ISBN 978-1-879284-67-8.
- Kabbani H, Raghuveer TS (June 2004). "Craniosynostosis". American Family Physician. 69 (12): 2863–70. PMID 15222651.
External links
- GeneReview/NIH/UW entry on FGFR-Related Craniosynostosis Syndromes
- Ebenezer, Roy (1960). "Craniostenosis or oxycephaly". Indian Journal of Ophthalmology. 8 (3): 77–80. ISSN 0301-4738. PMID 13819157.