Dentinogenesis imperfecta
Dentinogenesis imperfecta (DI) is a genetic disorder of tooth development. It is inherited in an autosomal dominant pattern, as a result of mutations on chromosome 4q21, in the dentine sialophosphoprotein gene (DSPP).[1][2][3][4][5] It is one of the most frequently occurring autosomal dominant features in humans.[6] Dentinogenesis imperfecta affects an estimated 1 in 6,000-8,000 people.[7]
| Dentinogenesis imperfecta | |
|---|---|
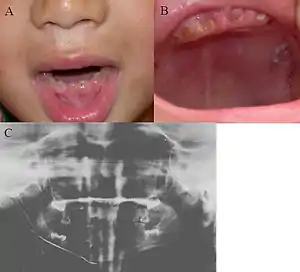 | |
| Oral photographs from an individual with Dentinogenesis imperfecta | |
| Specialty | Dentistry |
This condition can cause teeth to be discolored (most often a blue-gray or yellow-brown color) and translucent, giving teeth an opalescent sheen.[2][3][8][5][9] Teeth are also weaker than normal, making them prone to rapid wear, breakage, and loss.[2][3][4][5][8] These problems can affect baby (primary/deciduous) teeth alone, or both baby teeth and adult (permanent) teeth, with the baby teeth usually more severely affected.[5][8]
Although genetic factors are the main contributor for the disease, any environmental or systemic upset that impedes calcification or metabolisation of calcium can also result in anomalous dentine.
Classification
Shield classification (1973)
This is the most widely used classification for dentinogenesis imperfecta, and sub-divides the condition into 3 types:
Type I
DI associated with Osteogenesis Imperfecta (OI).[2][5][10] Type of DI with similar dental abnormalities usually an autosomal dominant trait with variable expressivity but can be recessive if the associated osteogenesis imperfecta is of recessive type.[11]
Recent genetic studies have identified that mutations in the genes coding for the collagen type 1 proteins, COL1A1 and COL1A2, are associated with this type of DI.[2][8]
Not all individuals with OI have dentinogenesis imperfecta, and the prevalence of DI varies depending on the sub-type of OI:
Type II
DI not associated with OI. Occurs in people without other inherited disorders (i.e. Osteogenesis imperfecta). It is an autosomal dominant trait. A few families with type II have progressive hearing loss in addition to dental abnormalities. Also called hereditary opalescent dentin.[12]
Type III
Brandywine isolate. This type is rare with occurrences only in the secluded populations in Maryland, USA.[3][13][14] Similar to DI type II, this type is also not associated with OI.[2] Its predominant characteristic is bell-shaped crowns, especially in the permanent dentition. Unlike Types I and II, it involves teeth with shell-like appearance and multiple pulp exposures.[12]
Mutations in the gene coding for the dentine sialophosphoprotein (DSPP) are associated with DI type II and III. DSPP is a polypeptide which gives rise to 3 proteins; dentine sialoprotein (DSP), dentine glycoprotein (DGP), and dentine phosphoprotein (DPP). The DPP protein is thought to contribute to hydroxyapatite crystal formation and growth, a fundamental crystal which is widely distributed in mineralised dentine and enamel. The function of the DGP and DSP proteins is not well understood.[2][3][4]
Genetic studies have shown that type II and III may be the same sub-type of dentinogenesis imperfecta, differing only by the severity.[4]
de La Dure-Molla, Foruner and Berdal (2015)
de La Dure-Molla, Foruner and Berdal (2015) have proposed a new classification to supersede the Shield Classification (1973). This new classification is designed to overcome the shortcomings of its predecessor, mainly the clinical difficulty in using the Shield classification due to the overlapping signs & symptoms between the sub-types.[3]
In this classification, the authors propose that the DSPP (dentine sialophosphoprotein) diseases, that is dentinogenesis imperfecta and dentine dysplasia, are jointly named "Dentinogenesis imperfecta", and sub-types are determined according to the severity of the condition. There are a few exceptions:
- Shields' Dentine Dysplasia type I - this condition is unique in that it only affects root development, and is separately termed "radicular dentin dysplasia" in the new classification.
- Shields' Dentinogenesis Imperfecta type I - this sub-type is not acknowledged in this new classification as the authors deem it a different disease since it is a syndrome of osteogenesis imperfecta[3]
Mild type
Primary (baby) teeth are moderately affected.
Permanent (adult) teeth are not discoloured, or the discolouration is mild (grey colour). Little or no attrition (tooth wear) is evident. The crown of the teeth may be bulbous and markedly constricted at the cemento-enamel junction (CEJ).
Radiographically, evidence of partial pulp obliteration with a "thistle-shaped appearance".[3]
Moderate type
Teeth are moderately discoloured (blue, grey or amber opalescent). More attrition is evident with shortening of crown height. Crowns may appear bulbous with prominent constriction at the CEJ.
Radiographically, the pulp is small or is totally obliterated. Roots appear thinner and shorter than average. There may be periapical pathology.[3]
Severe type
Teeth are markedly discoloured (brown opalescent). The crowns are very short due to severe attrition. Crowns may appear bulbous with prominent constriction at the CEJ.
Radiographically, pulp appears large and the dentine layer is thin ("shell teeth" as described in Presentation section). Roots are thin and short. There may be multiple periapical pathologies.[3]
Radicular dentin dysplasia
This sub-type is used in place of Shields' dentine dysplasia type I, in which only the roots of the teeth are affected.
Both primary and permanent teeth are affected.
The teeth appear normal clinically. Radiographically, the roots are shorter and fused together with a rounded apex.[3]
Presentation
Clinical presentation
Clinical features include:
- Discoloured teeth - teeth may be amber, brown, blue or opalescent
- Bulbous shape to the tooth crown due to cervical constriction
- Tooth wear/Non-carious tooth surface loss (NCTSL) - due to the poorly mineralised dentine, the enamel of the tooth is unsupported and subsequently shears or chips off as it is subjected to biting forces. This exposes the underlying poorly mineralised dentine which is less resistant to wear. Therefore, features of abrasion and attrition may become apparent.
- Reduction in occlusal vertical dimension (OVD) - this is secondary to the tooth wear/NCTSL. A reduced OVD can lead to craniofacial dysgnathia, poor tooth aesthetics, and disorders during chewing, swallowing, speaking and eating.[2][3][4][5][8]
The baby (primary) teeth are usually more severely affected than adult (permanent) teeth.[3][5][8]
Enamel is usually lost early because it is further inclined to attrition due to loss of scalloping at the dentinoenamel junction (DEJ). It was suggested that the scalloping is beneficial for the mechanical properties of teeth as it reinforces the anchor between enamel and dentine.[15] However, the teeth are not more susceptible to dental caries than normal ones.
Periodontal disease, or gum disease, is a common finding amongst individuals with dentinogenesis imperfecta despite no clinical findings of tooth decay (dental caries). The reason for this is currently not well understood.[3]
Certain patients with dentinogenesis imperfecta will suffer from multiple periapical abscesses apparently resulting from pulpal strangulation secondary to pulpal obliteration or from pulp exposure due to extensive coronal wear. They may need apical surgery to save the involved teeth.[12]
Note that, although dentine exposure is a common clinical finding, individuals with dentinogenesis imperfecta usually do not experience tooth sensitivity as the exposed dentine is typically sclerosed (hardened), thereby appearing glassy/shiny.[2]
Radiographic presentation
Radiographic features include:
- Bulbous shape of tooth crown with pronounced cervical constriction
- Small pulp, or total pulp obliteration
- Small or obliterated root canal
- Presence of pulp stones
- Narrow and small roots
- Periapical radiolucency without any evidence of clinical pathology such as tooth decay (dental caries)[2][3][5][14]
Presentation by sub-type of Dentinogenesis Imperfecta
Clinical and radiographic features can be categorised by the sub-type of dentinogenesis imperfecta (see Shield's Classification in the Classification section):
Type I
Clinically, both the baby (primary) and adult (permanent) teeth often appear amber coloured and translucent, and show signs of severe attrition. Primary teeth have a more obvious appearance as they have a thinner layer of enamel overlying dentine, hence the abnormal color of dentine is more noticeable.[2]
Radiographically, affected teeth have short and narrow roots, and obliterated pulps due to dentine hypertrophy before or shortly after tooth eruption.[2]
The severity of these features is variable, with some teeth presenting with total obliteration of the pulp, while other teeth appear to have normal, healthy dentine.[2]
Some type I cases present with no clinical findings, with only radiographic abnormalities.[4]
Type II
Type II has a similar clinical and radiographic appearance to type I[16] with some distinguishing features:
- Bulbous crowns are common with pronounced cervical constriction
- All teeth in the mouth are affected, with severe abnormalities present in both the baby (primary) and adult (permanent) teeth. This is in contrast to type I where the presentation is more variable
- Rarely, individuals exhibit sensorineural hearing loss. It is proposed this hearing loss is a secondary feature to attrition; this type of tooth wear can cause jaw overclosure with subsequent changes to the shape of the inner ear, thus causing hearing loss. However, the true cause remains unknown.[2][4]
Type III
Similar clinical and radiographic features to that of type I and II are apparent for the adult (permanent) teeth. The main distinguishing feature is "shell teeth", a term used to describe the unique appearance of the baby (primary) teeth; the primary teeth have multiple pulp exposures and radiographically appear hollow as the dentine layer is thin (dentine hypotrophy) and the pulp chamber is very large.[2][3][4][14]
Histology
The enamel has a regular structure, however, there are abnormalities in the structure of dentine and at the amelo-dentinal junction.[4][5] These abnormalities include:
Diagnosis
To determine if the condition has been inherited, it is suggested to ask if any other family member has Dentinogenesis imperfecta. A lack of family history may indicate that the condition was acquired.[2]
It is suggested that the dental/medical professional establish if the condition is a syndrome of another inherited condition such as:
- Osteogenesis imperfecta - indicates Type I Dentinogenesis Imperfecta (see Shield's Classification in Classification section)
- Ehlers Danlos syndrome
- Goldblatt syndrome
- Schimke immune-osseus dysplasia
- Brachio-skeleto-genital syndrome
- Osteodysplastic and primordial short stature with severe microdontia
- Opalescent teeth
- Rootless molars[2]
[This is not a comprehensive list]
It can be useful to enquire about symptoms of osteogenesis imperfecta, as Type I Dentinogenesis Imperfecta (Shield's Classification) is associated with osteogenesis imperfecta.[2] Notable information includes:
Common dental features of osteogenesis imperfecta include:
- Hypodontia
- Oligodontia
- Taurodontism
- Unerupted permanent 2nd molars, with no obstruction in the path of eruption
- Retrognathic maxilla[10][17][18]
Differential diagnosis
- Hypocalcified forms of amelogenesis imperfecta
- Congenital erythropoietic porphyria
- Conditions that can cause early loss of teeth e.g. Kostmann's disease, cyclic neutropenia, Chediak-Hegashi syndrome, histiocytosis X, Papillon-Lefevre syndrome
- Permanent tooth discolouration caused by medications such as tetracyclines, or medical conditions such as rickets[2]
Treatment
Preventive and restorative care are important as well as esthetics as a consideration. This ensures preservation of the patient's vertical face height between their upper and lower teeth when they bite together. The basis of treatment is standard throughout the different types of DI where prevention, preservation of occlusal face height, maintenance of function, and aesthetic needs are priority. Preventive efforts can limit pathology occurring within the pulp, which may render future endodontic procedures less challenging, with better outcomes.
- Challenges are associated with root canal treatment of teeth affected by DI due to pulp chamber and root canal obliteration, or narrowing of such spaces.
- If root canal treatment is indicated, it should be done in a similar way like with any other tooth.[19] Further consideration is given for restoring the root-treated tooth as it has weaker dentine which may not withstand the restoration.
Preservation of occlusal face height may be tackled by use of stainless steel crowns which are advocated for primary teeth where occlusal face height may be hugely compromised due to loss of tooth tissue as a result of attrition, erosion of enamel.[14]
In most cases, full-coverage crowns or veneers (composite/porcelain) are needed for aesthetic appearance, as well as to prevent further attrition.[9] Another treatment option is bonding, putting lighter enamel on the weakened enamel of the teeth and with many treatments of this bonding, the teeth appear whiter to the eye, but the teeth on the inside and under that cover are still the same. Due to the weakened condition of the teeth, many common cosmetic procedures such as braces and bridges are inappropriate for patients with Dentinogenesis imperfecta and are likely to cause even more damage than the situation they were intended to correct.
Dental whitening (bleaching) is contraindicated although it has been reported to lighten the color of DI teeth with some success; however, because the discoloration is caused primarily by the underlying yellow-brown dentin, this alone is unlikely to produce normal appearance in cases of significant discoloration.[12]
If there is considerable attrition, overdentures may be prescribed to prevent further attrition of remaining teeth and for preserving the occlusal face height.[14]
Management of DI associated with OI
Bisphosphonates have recently been introduced to treat several bone disorders, which include osteogenesis imperfecta.
A recognized risk of this drug relevant to dental treatments is bisphosphonate-associated osteonecrosis of the jaw (BRONJ).[20][21] Occurrences of this risk is associated with dental surgical procedures such as extractions.
Dental professionals should therefore proceed with caution when carrying out any dental procedures in patients who have Type 2 DI who may be on bisphosphonate drug therapy.
References
- Beattie ML, Kim JW, Gong SG, Murdoch-Kinch CA, Simmer JP, Hu JC (April 2006). "Phenotypic variation in dentinogenesis imperfecta/dentin dysplasia linked to 4q21". Journal of Dental Research. 85 (4): 329–333. doi:10.1177/154405910608500409. PMC 2238637. PMID 16567553.
- Barron MJ, McDonnell ST, Mackie I, Dixon MJ (November 2008). "Hereditary dentine disorders: dentinogenesis imperfecta and dentine dysplasia". Orphanet Journal of Rare Diseases. 3 (1): 31. doi:10.1186/1750-1172-3-31. PMC 2600777. PMID 19021896.
- de La Dure-Molla M, Philippe Fournier B, Berdal A (April 2015). "Isolated dentinogenesis imperfecta and dentin dysplasia: revision of the classification". European Journal of Human Genetics. 23 (4): 445–451. doi:10.1038/ejhg.2014.159. PMC 4666581. PMID 25118030.
- Soliman S, Meyer-Marcotty P, Hahn B, Halbleib K, Krastl G (September 2018). "Treatment of an Adolescent Patient with Dentinogenesis Imperfecta Using Indirect Composite Restorations - A Case Report and Literature Review". The Journal of Adhesive Dentistry. 20 (4): 345–354. doi:10.3290/j.jad.a40991. PMID 30206577.
- Malmgren B, Norgren S (March 2002). "Dental aberrations in children and adolescents with osteogenesis imperfecta". Acta Odontologica Scandinavica. 60 (2): 65–71. doi:10.1080/000163502753509446. PMID 12020117. S2CID 110970.
- Thotakura SR, Mah T, Srinivasan R, Takagi Y, Veis A, George A (March 2000). "The non-collagenous dentin matrix proteins are involved in dentinogenesis imperfecta type II (DGI-II)". Journal of Dental Research. 79 (3): 835–839. doi:10.1177/00220345000790030901. PMID 10765957. S2CID 38418321.
- Witkop CJ (January 1975). "Hereditary defects of dentin". Dental Clinics of North America. 19 (1): 25–45. doi:10.1016/S0011-8532(22)00655-3. PMID 162890. S2CID 22518085.
- Ma MS, Najirad M, Taqi D, Retrouvey JM, Tamimi F, Dagdeviren D, et al. (March 2019). "Caries prevalence and experience in individuals with osteogenesis imperfecta: A cross-sectional multicenter study". Special Care in Dentistry. 39 (2): 214–219. doi:10.1111/scd.12368. PMC 6402806. PMID 30758072.
- Bath-Balogh M (2011). Illustrated Dental Embryology, Histology, and Anatomy (3rd ed.). Elsevier Health Sciences. p. 64. ISBN 978-0-323-29086-9.
- Andersson K, Dahllöf G, Lindahl K, Kindmark A, Grigelioniene G, Åström E, Malmgren B (2017-05-12). Divaris K (ed.). "Mutations in COL1A1 and COL1A2 and dental aberrations in children and adolescents with osteogenesis imperfecta - A retrospective cohort study". PLOS ONE. 12 (5): e0176466. Bibcode:2017PLoSO..1276466A. doi:10.1371/journal.pone.0176466. PMC 5428910. PMID 28498836.
- Nanci A (2013). Ten Cate's oral histology: development, structure, and function (8th ed.). St. Louis, Mo.: Elsevier. p. 15. ISBN 978-0-323-07846-7.
- Guideline on Dental Management of Heritable Dental Developmental Anomalies (PDF). American Academy of Pediatric Dentistry. 2013.
- Huth KC, Paschos E, Sagner T, Hickel R (September 2002). "Diagnostic features and pedodontic-orthodontic management in dentinogenesis imperfecta type II: a case report". International Journal of Paediatric Dentistry. 12 (5): 316–321. doi:10.1046/j.1365-263X.2002.00390.x. PMID 12199890.
- Pettiette MT, Wright JT, Trope M (December 1998). "Dentinogenesis imperfecta: endodontic implications. Case report". Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 86 (6): 733–737. doi:10.1016/s1079-2104(98)90213-x. PMID 9868734.
- Shimizu D, Macho GA (January 2007). "Functional significance of the microstructural detail of the primate dentino-enamel junction: a possible example of exaptation". Journal of Human Evolution. 52 (1): 103–111. doi:10.1016/j.jhevol.2006.08.004. PMID 16997355.
- Rios D, Falavinha A, Tenuta L, Machado M (2005). "Osteogenesis imperfecta and dentinogenesis imperfecta: associated disorders". Quintessence International. 36 (9): 695–701. PMID 16163872.
- Jensen BL, Lund AM (1997-07-01). "Osteogenesis imperfecta: clinical, cephalometric, and biochemical investigations of OI types I, III, and IV". Journal of Craniofacial Genetics and Developmental Biology. 17 (3): 121–132. PMID 9338855.
- Waltimo-Sirén J, Kolkka M, Pynnönen S, Kuurila K, Kaitila I, Kovero O (March 2005). "Craniofacial features in osteogenesis imperfecta: a cephalometric study". American Journal of Medical Genetics. Part A. 133A (2): 142–150. doi:10.1002/ajmg.a.30523. PMID 15666304. S2CID 23172499.
- Henke DA, Fridrich TA, Aquilino SA (May 1999). "Occlusal rehabilitation of a patient with dentinogenesis imperfecta: a clinical report". The Journal of Prosthetic Dentistry. 81 (5): 503–506. doi:10.1016/s0022-3913(99)70201-5. PMID 10220651.
- Woo SB, Hellstein JW, Kalmar JR (May 2006). "Narrative [corrected] review: bisphosphonates and osteonecrosis of the jaws". Annals of Internal Medicine. 144 (10): 753–761. doi:10.7326/0003-4819-144-10-200605160-00009. PMID 16702591. S2CID 53091343.
- Khosla S, Burr D, Cauley J, Dempster DW, Ebeling PR, Felsenberg D, et al. (October 2007). "Bisphosphonate-associated osteonecrosis of the jaw: report of a task force of the American Society for Bone and Mineral Research". Journal of Bone and Mineral Research. 22 (10): 1479–1491. doi:10.1359/jbmr.0707onj. PMID 17663640.
This article incorporates public domain text from The U.S. National Library of Medicine