Gardner's syndrome
Gardner's syndrome (also known as Gardner syndrome, familial polyposis of the colon,[1] or familial colorectal polyposis[2]) is a subtype of familial adenomatous polyposis (FAP). Gardner syndrome is an autosomal dominant form of polyposis characterized by the presence of multiple polyps in the colon together with tumors outside the colon.[3] The extracolonic tumors may include osteomas of the skull, thyroid cancer, epidermoid cysts, fibromas,[4] as well as the occurrence of desmoid tumors in approximately 15% of affected individuals.
| Gardner's syndrome | |
|---|---|
| Other names | Familial colorectal polyposis, Familial polyposis of the colon |
 | |
| Specialty | Gastroenterology, oncology, medical genetics |
Desmoid tumors are fibrous tumors that usually occur in the tissue covering the intestines and may be provoked by surgery to remove the colon. The countless polyps in the colon predispose to the development of colon cancer; if the colon is not removed, the chance of colon cancer is considered to be very significant. Polyps may also grow in the stomach, duodenum, spleen, kidneys, liver, mesentery, and small bowel. In a small number of cases, polyps have also appeared in the cerebellum. Cancers related to Gardner syndrome commonly appear in the thyroid, liver and kidneys. The number of polyps increases with age, and hundreds to thousands of polyps can develop in the colon.
The syndrome was first described in 1951.[5] There is no cure at this time, and in its more advanced forms, it is considered a terminal diagnosis with a life expectancy of 35–45 years; treatments are surgery and palliative care, although some chemotherapy has been tried with limited success.[6]
Cause
Gardner syndrome is caused by mutation in the adenomatous polyposis coli (APC gene), located in chromosome 5q21 (band q21 on chromosome 5).[3] This gene is also mutant in familial adenomatous polyposis (FAP), a more common disease that also predisposes to colon cancer. Nuances in the understanding of genetics have caused some disorders to be split into multiple entities, while others merged into one genetic condition. After most of the second half of the 20th century, Gardner syndrome has been merged into FAP and is now considered simply a phenotypic subtype of FAP.[7] FAP defined by the development of hundreds or thousands of polyps in the colon. Gardner syndrome is set apart as a subtype because, in addition to colonic polyps, there are also extra-colonic growths (both malignant and benign).[8] There are many terms used to describe "APC-associated polyposis condition" including FAP, attenuated FAP, Gardner syndrome, Turcot syndrome, and gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS). There is a movement toward no longer using the terms Gardner Syndrome or Turcot Syndrome since both are part of the FAP spectrum. Gardner syndrome and Turcot syndrome are regarded primarily for historical interest.[9]
Genetics
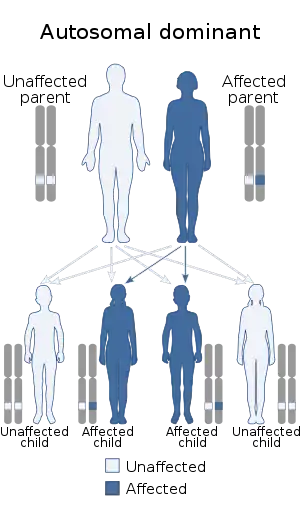
Gardner syndrome is inherited in an autosomal dominant manner.[3] Typically, one parent has Gardner syndrome. Each of their children, male and female alike, are at 50% risk of inheriting the gene for Gardner syndrome.
Diagnosis
Gardner syndrome consists of adenomatous polyps of the gastrointestinal tract, Gardner fibromas, desmoid tumors, osteomas, epidermoid cysts, lipomas, dental abnormalities, and periampullary carcinomas. The incidence of the syndrome is 1:14,025 with an equal sex distribution. It is determined by the autosomal dominant familial polyposis coli gene (APC) on chromosome 5.[5][10]
Gardner syndrome can be identified based on oral findings, including multiple impacted and supernumerary teeth, multiple jaw osteomas that give a "cotton-wool" appearance to the jaws, as well as multiple odontomas, congenital hypertrophy of the retinal pigment epithelium (CHRPE), in addition to multiple adenomatous polyps of the colon. Gardner syndrome is also associated with familial adenomatous polyposis and may manifest as aggressive fibromatosis (desmoid tumors) of the retroperitoneum.[11]
Desmoid tumors arise most frequently from the aponeurosis of the rectus abdominal muscle of multiparous women. The extra-abdominal form is rare and desmoids of the breast may arise in the mammary gland or may occur as an extension of a lesion arising from the muscles of the chest wall. The incidence of mammary desmoid tumors is less than 0.2% of primary breast neoplasms. In Gardner's syndrome, the incidence ranges from 4% to 17%. Desmoid tumors associated with Gardner's syndrome have been shown to have an alteration of the β-catenin pathway and over express β-catenin.[5]
Treatment
There is no cure for Gardner Syndrome. Treatments focus on alleviating symptoms and reducing risk of cancer. Treatments for desmoid tumors may include surgery, NSAIDS, anti-estrogen medications, radiation therapy and chemotherapy.[6]
Eponym
The syndrome is named for Eldon J. Gardner (1909–1989), a geneticist who first described it in 1951.[12]
See also
References
- Dermatology. Bolognia, Jean,, Schaffer, Julie V.,, Cerroni, Lorenzo (Fourth ed.). [Philadelphia, Pa.] 22 October 2017. p. 1031. ISBN 978-0-7020-6342-8. OCLC 1011508489.
{{cite book}}: CS1 maint: others (link) - Dermatology. Bolognia, Jean,, Schaffer, Julie V.,, Cerroni, Lorenzo (Fourth ed.). [Philadelphia, Pa.] 22 October 2017. p. 1223. ISBN 978-0-7020-6342-8. OCLC 1011508489.
{{cite book}}: CS1 maint: others (link) - Online Mendelian Inheritance in Man (OMIM): 175100
- Luba MC, Bangs SA, Mohler AM, Stulberg DL (February 2003). "Common benign skin tumors". Am Fam Physician. 67 (4): 729–38. PMID 12613727. Archived from the original on 2008-05-16. Retrieved 2009-08-23.
- Rammohan A, Wood JJ (2012). "Desmoid tumour of the breast as a manifestation of Gardner's syndrome". Int J Surg Case Rep. 3 (5): 139–142. doi:10.1016/j.ijscr.2012.01.004. PMC 3312056. PMID 22370045.
- "Gardner syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 17 April 2018.
- "Gardner Syndrome". Cancer.net. American Society of Clinical Oncology. 2012-06-26. Retrieved 8 July 2016.
- "Gardner syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2018-09-20.
- "Gardner syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2018-09-20.
- Baranov E, Hornick JL (March 2020). "Soft Tissue Special Issue: Fibroblastic and Myofibroblastic Neoplasms of the Head and Neck". Head and Neck Pathology. 14 (1): 43–58. doi:10.1007/s12105-019-01104-3. PMC 7021862. PMID 31950474.
- DeVita. Cancer, Principles and Practice of Oncology, 8th Ed. p. 1742.
- Gardner EJ (June 1951). "A genetic and clinical study of intestinal polyposis, a predisposing factor for carcinoma of the colon and rectum". Am. J. Hum. Genet. 3 (2): 167–76. PMC 1716321. PMID 14902760.
External links
- Gardner syndrome at NIH's Office of Rare Diseases