JAK-STAT signaling pathway
The JAK-STAT signaling pathway is a chain of interactions between proteins in a cell, and is involved in processes such as immunity, cell division, cell death, and tumour formation. The pathway communicates information from chemical signals outside of a cell to the cell nucleus, resulting in the activation of genes through the process of transcription. There are three key parts of JAK-STAT signalling: Janus kinases (JAKs), signal transducer and activator of transcription proteins (STATs), and receptors (which bind the chemical signals).[1] Disrupted JAK-STAT signalling may lead to a variety of diseases, such as skin conditions, cancers, and disorders affecting the immune system.[1]
Structure of JAKs and STATs
Main articles: JAKs and STATs
There are four JAK proteins: JAK1, JAK2, JAK3 and TYK2.[1] JAKs contains a FERM domain (approximately 400 residues), an SH2-related domain (approximately 100 residues), a kinase domain (approximately 250 residues) and a pseudokinase domain (approximately 300 residues).[2] The kinase domain is vital for JAK activity, since it allows JAKs to phosphorylate (add phosphate groups to) proteins.
There are seven STAT proteins: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B and STAT6.[1] STAT proteins contain many different domains, each with a different function, of which the most conserved region is the SH2 domain.[2] The SH2 domain is formed of 2 α-helices and a β-sheet and is formed approximately from residues 575–680.[2][3] STATs also have transcriptional activation domains (TAD), which are less conserved and are located at the C-terminus.[4] In addition, STATs also contain: tyrosine activation, amino-terminal, linker, coiled-coil and DNA-binding domains.[4]
Mechanism

The binding of various ligands, usually cytokines, such as interferons and interleukins, to cell-surface receptors, causes the receptors to dimerize, which brings the receptor-associated JAKs into close proximity.[6] The JAKs then phosphorylate each other on tyrosine residues located in regions called activation loops, through a process called transphosphorylation, which increases the activity of their kinase domains.[6] The activated JAKs then phosphorylate tyrosine residues on the receptor, creating binding sites for proteins possessing SH2 domains.[6] STATs then bind to the phosphorylated tyrosines on the receptor using their SH2 domains, and then they are tyrosine-phosphorylated by JAKs, causing the STATs to dissociate from the receptor.[2] At least STAT5 requires glycosylation at threonine 92 for strong STAT5 tyrosine phosphorylation.[7] These activated STATs form hetero- or homodimers, where the SH2 domain of each STAT binds the phosphorylated tyrosine of the opposite STAT, and the dimer then translocates to the cell nucleus to induce transcription of target genes.[2] STATs may also be tyrosine-phosphorylated directly by receptor tyrosine kinases - but since most receptors lack built-in kinase activity, JAKs are usually required for signalling.[1]
Movement of STATs from the cytosol to the nucleus
To move from the cytosol to the nucleus, STAT dimers have to pass through nuclear pore complexes (NPCs), which are protein complexes present along the nuclear envelope that control the flow of substances in and out of the nucleus. To enable STATs to move into the nucleus, an amino acid sequence on STATs, called the nuclear localization signal (NLS), is bound by proteins called importins.[4] Once the STAT dimer (bound to importins) enters the nucleus, a protein called Ran (associated with GTP) binds to the importins, releasing them from the STAT dimer.[8] The STAT dimer is then free in the nucleus.
Specific STATs appear to bind to specific importin proteins. For example, STAT3 proteins can enter the nucleus by binding to importin α3 and importin α6.[9] On the other hand, STAT1 and STAT2 bind to importin α5.[4] Studies indicate that STAT2 requires a protein called interferon regulatory factor 9 (IRF9) to enter the nucleus.[8] Not as much is known about nuclear entrance of other STATs, but it has been suggested that a sequence of amino acids in the DNA-binding domain of STAT4 might allow nuclear import; also, STAT5 and STAT6 can both bind to importin α3.[8] In addition, STAT3, STAT5 and STAT6 can enter the nucleus even if they are not phosphorylated at tyrosine residues.[8]
Role of post-translational modifications
After STATs are made by protein biosynthesis, they have non-protein molecules attached to them, called post-translational modifications. One example of this is tyrosine phosphorylation (which is fundamental for JAK-STAT signalling), but STATs experience other modifications, which may affect STAT behaviour in JAK-STAT signalling. These modifications include: methylation, acetylation and serine phosphorylation.
- Methylation. STAT3 can be dimethylated (have two methyl groups) on a lysine residue, at position 140, and it is suggested that this could reduce STAT3 activity.[10] There is debate as to whether STAT1 is methylated on an arginine residue (at position 31), and what the function of this methylation could be.[11]
- Acetylation. STAT1, STAT2, STAT3, STAT5 and STAT6 have been shown to be acetylated.[12] STAT1 may have an acetyl group attached to lysines at positions 410 and 413, and as a result, STAT1 can promote the transcription of apoptotic genes - triggering cell death.[12] STAT2 acetylation is important for interactions with other STATs, and for the transcription of anti-viral genes.[4]
Acetylation of STAT3 has been suggested to be important for its dimerization, DNA-binding and gene-transcribing ability, and IL-6 JAK-STAT pathways that use STAT3 require acetylation for transcription of IL-6 response genes.[12] STAT5 acetylation on lysines at positions 694 and 701 is important for effective STAT dimerization in prolactin signalling.[13] Adding acetyl groups to STAT6 is suggested to be essential for gene transcription in some forms of IL-4 signalling, but not all the amino acids which are acetylated on STAT6 are known.[12]
- Serine phosphorylation. Most of the seven STATs (except STAT2) undergo serine phosphorylation.[2] Serine phosphorylation of STATs has been shown to reduce gene transcription.[14] It is also required for the transcription of some target genes of the cytokines IL-6 and IFN- γ.[11] It has been proposed that phosphorylation of serine can regulate STAT1 dimerization,[11] and that continuous serine phosphorylation on STAT3 influences cell division.[15]
Recruitment of co-activators
Like many other transcription factors, STATs are capable of recruiting co-activators such as CBP and p300, and these co-activators increase the rate of transcription of target genes.[2] The coactivators are able to do this by making genes on DNA more accessible to STATs and by recruiting proteins needed for transcription of genes. The interaction between STATs and coactivators occurs through the transactivation domains (TADs) of STATs.[2] The TADs on STATs can also interact with histone acetyltransferases (HATs);[16] these HATs add acetyl groups to lysine residues on proteins associated with DNA called histones. Adding acetyl groups removes the positive charge on lysine residues, and as a result there are weaker interactions between histones and DNA, making DNA more accessible to STATs and enabling an increase in the transcription of target genes.
Integration with other signalling pathways
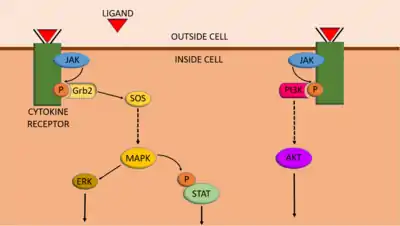
JAK-STAT signalling is able to interconnect with other cell-signalling pathways, such as the PI3K/AKT/mTOR pathway.[17] When JAKs are activated and phosphorylate tyrosine residues on receptors, proteins with SH2 domains (such as STATs) are able bind to the phosphotyrosines, and the proteins can carry out their function. Like STATs, the PI3K protein also has an SH2 domain, and therefore it is also able to bind to these phosphorylated receptors.[17] As a result, activating the JAK-STAT pathway can also activate PI3K/AKT/mTOR signalling.
JAK-STAT signalling can also integrate with the MAPK/ERK pathway. Firstly, a protein important for MAPK/ERK signalling, called Grb2, has an SH2 domain, and therefore it can bind to receptors phosphorylated by JAKs (in a similar way to PI3K).[17] Grb2 then functions to allow the MAPK/ERK pathway to progress. Secondly, a protein activated by the MAPK/ERK pathway, called MAPK (mitogen-activated protein kinase), can phosphorylate STATs, which can increase gene transcription by STATs.[17] However, although MAPK can increase transcription induced by STATs, one study indicates that phosphorylation of STAT3 by MAPK can reduce STAT3 activity.[18]
One example of JAK-STAT signalling integrating with other pathways is Interleukin-2 (IL-2) receptor signaling in T cells. IL-2 receptors have γ (gamma) chains, which are associated with JAK3, which then phosphorylates key tyrosines on the tail of the receptor.[19] Phosphorylation then recruits an adaptor protein called Shc, which activates the MAPK/ERK pathway, and this facilitates gene regulation by STAT5.[19]
Alternative signalling pathway
An alternative mechanism for JAK-STAT signalling has also been suggested. In this model, SH2 domain-containing kinases, can bind to phosphorylated tyrosines on receptors and directly phosphorylate STATs, resulting in STAT dimerization.[6] Therefore, unlike the traditional mechanism, STATs can be phosphorylated not just by JAKs, but by other receptor-bound kinases. So, if one of the kinases (either JAK or the alternative SH2-containing kinase) cannot function, signalling may still occur through activity of the other kinase.[6] This has been shown experimentally.[20]
Role in cytokine receptor signalling
Given that many JAKs are associated with cytokine receptors, the JAK-STAT signalling pathway plays a major role in cytokine receptor signalling. Since cytokines are substances produced by immune cells that can alter the activity of neighbouring cells, the effects of JAK-STAT signalling are often more highly seen in cells of the immune system. For example, JAK3 activation in response to IL-2 is vital for lymphocyte development and function.[21] Also, one study indicates that JAK1 is needed to carry out signalling for receptors of the cytokines IFNγ, IL-2, IL-4 and IL-10.[22]
The JAK-STAT pathway in cytokine receptor signalling can activate STATs, which can bind to DNA and allow the transcription of genes involved in immune cell division, survival, activation and recruitment. For example, STAT1 can enable the transcription of genes which inhibit cell division and stimulate inflammation.[2] Also, STAT4 is able to activate NK cells (natural killer cells), and STAT5 can drive the formation of white blood cells.[2][23] In response to cytokines, such as IL-4, JAK-STAT signalling is also able to stimulate STAT6, which can promote B-cell proliferation, immune cell survival, and the production of an antibody called IgE.[2]
Role in development
JAK-STAT signalling plays an important role in animal development. The pathway can promote blood cell division, as well as differentiation (the process of a cell becoming more specialised).[24] In some flies with faulty JAK genes, too much blood cell division can occur, potentially resulting in leukaemia.[25] JAK-STAT signalling has also been associated with excessive white blood cell division in humans and mice.[24]
The signalling pathway is also crucial for eye development in the fruit fly (Drosophila melanogaster). When mutations occur in genes coding for JAKs, some cells in the eye may be unable to divide, and other cells, such as photoreceptor cells, have been shown not to develop correctly.[24]
The entire removal of a JAK and a STAT in Drosophila causes death of Drosophila embryos, whilst mutations in the genes coding for JAKs and STATs can cause deformities in the body patterns of flies, particularly defects in forming body segments.[24] One theory as to how interfering with JAK-STAT signalling might cause these defects is that STATs may directly bind to DNA and promote the transcription of genes involved in forming body segments, and therefore by mutating JAKs or STATs, flies experience segmentation defects.[26] STAT binding sites have been identified on one of these genes, called even-skipped (eve), to support this theory.[27] Of all the segment stripes affected by JAK or STAT mutations, the fifth stripe is affected the most, the exact molecular reasons behind this are still unknown.[24]
Regulation
Given the importance of the JAK-STAT signalling pathway, particularly in cytokine signalling, there are a variety of mechanisms that cells possess to regulate the amount of signalling that occurs. Three major groups of proteins that cells use to regulate this signalling pathway are protein inhibitors of activated STAT (PIAS),[28] protein tyrosine phosphatases (PTPs) [29] and suppressors of cytokine signalling (SOCS).[30] Computational models of JAK-STAT signaling based on the laws of chemical kinetics have elucidated the importance of these different regulatory mechanisms on JAK-STAT signaling dynamics.[31][32][33]
Protein inhibitors of activated STATs (PIAS)
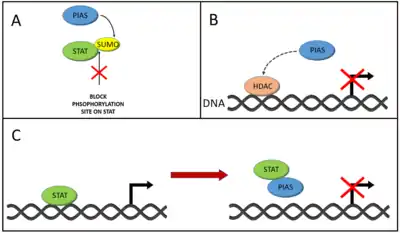
PIAS are a four-member protein family made of: PIAS1, PIAS3, PIASx, and PIASγ.[34] The proteins add a marker, called SUMO (small ubiquitin-like modifier), onto other proteins – such as JAKs and STATs, modifying their function.[34] The addition of a SUMO group onto STAT1 by PIAS1 has been shown to prevent activation of genes by STAT1.[35] Other studies have demonstrated that adding a SUMO group to STATs may block phosphorylation of tyrosines on STATs, preventing their dimerization and inhibiting JAK-STAT signalling.[36] PIASγ has also been shown to prevent STAT1 from functioning.[37] PIAS proteins may also function by preventing STATs from binding to DNA (and therefore preventing gene activation), and by recruiting proteins called histone deacetylases (HDACs), which lower the level of gene expression.[34]
Protein tyrosine phosphatases (PTPs)
Since adding phosphate groups on tyrosines is such an important part of how the JAK-STAT signalling pathway functions, removing these phosphate groups can inhibit signalling. PTPs are tyrosine phosphatases, so are able to remove these phosphates and prevent signalling. Three major PTPs are SHP-1, SHP-2 and CD45.[38]
- SHP-1. SHP-1 is mainly expressed in blood cells.[39] It contains two SH2 domains and a catalytic domain (the region of a protein that carries out the main function of the protein) - the catalytic domain contains the amino acid sequence VHCSAGIGRTG (a sequence typical of PTPs).[40] As with all PTPs, a number of amino acid structures are essential for their function: conserved cysteine, arginine and glutamine amino acids, and a loop made of tryptophan, proline and aspartate amino acids (WPD loop).[40] When SHP-1 is inactive, the SH2 domains interact with the catalytic domain, and so the phosphatase is unable to function.[40] When SHP-1 is activated however, the SH2 domains move away from the catalytic domain, exposing the catalytic site and therefore allowing phosphatase activity.[40] SHP-1 is then able to bind and remove phosphate groups from the JAKs associated with receptors, preventing the transphosphorylation needed for the signalling pathway to progress.
One example of this is seen in the JAK-STAT signalling pathway mediated by the erythropoietin receptor (EpoR). Here, SHP-1 binds directly to a tyrosine residue (at position 429) on EpoR and removes phosphate groups from the receptor-associated JAK2.[41] The ability of SHP-1 to negatively regulate the JAK-STAT pathway has also been seen in experiments using mice lacking SHP-1.[42] These mice experience characteristics of autoimmune diseases and show high levels of cell proliferation, which are typical characteristics of an abnormally high level of JAK-STAT signalling.[42] Additionally, adding methyl groups to the SHP-1 gene (which reduces the amount of SHP-1 produced) has been linked to lymphoma (a type of blood cancer) .[43]
However, SHP-1 may also promote JAK-STAT signalling. A study in 1997 found that SHP-1 potentially allows higher amounts of STAT activation, as opposed to reducing STAT activity.[44] A detailed molecular understanding for how SHP-1 can both activate and inhibit the signalling pathway is still unknown.[38]
- SHP-2. SHP-2 has a very similar structure to SHP-1, but unlike SHP-1, SHP-2 is produced in many different cell types - not just blood cells.[45] Humans have two SHP-2 proteins, each made up of 593 and 597 amino acids.[40] The SH2 domains of SHP-2 appear to play an important role in controlling the activity of SHP-2. One of the SH2 domains binds to the catalytic domain of SHP-2, to prevent SHP-2 functioning.[38] Then, when a protein with a phosphorylated tyrosine binds, the SH2 domain changes orientation and SHP-2 is activated.[38] SHP-2 is then able to remove phosphate groups from JAKs, STATs and the receptors themselves - so, like SHP-1, can prevent the phosphorylation needed for the pathway to continue, and therefore inhibit JAK-STAT signalling. Like SHP-1, SHP-2 is able to remove these phosphate groups through the action of the conserved cysteine, arginine, glutamine and WPD loop.[40]
Negative regulation by SHP-2 has been reported in a number of experiments - one example has been when exploring JAK1/STAT1 signalling, where SHP-2 is able to remove phosphate groups from proteins in the pathway, such as STAT1.[46] In a similar manner, SHP-2 has also been shown to reduce signalling involving STAT3 and STAT5 proteins, by removing phosphate groups.[47][48]
Like SHP-1, SHP-2 is also believed to promote JAK-STAT signalling in some instances, as well as inhibit signalling. For example, one study indicates that SHP-2 may promote STAT5 activity instead of reducing it.[49] Also, other studies propose that SHP-2 may increase JAK2 activity, and promote JAK2/STAT5 signalling.[50] It is still unknown how SHP2 can both inhibit and promote JAK-STAT signalling in the JAK2/STAT5 pathway; one theory is that SHP-2 may promote activation of JAK2, but inhibit STAT5 by removing phosphate groups from it.[38]
- CD45. CD45 is mainly produced in blood cells.[4] In humans it has been shown to be able to act on JAK1 and JAK3,[51] whereas in mice, CD45 is capable of acting on all JAKs.[52] One study indicates that CD45 can reduce the amount of time that JAK-STAT signalling is active.[52] The exact details of how CD45 functions is still unknown.[38]
Suppressors of cytokine signalling (SOCS)
There are eight protein members of the SOCS family: cytokine-inducible SH2 domain-containing protein (CISH), SOCS1, SOCS2, SOCS3, SOCS4, SOCS5, SOCS6, and SOCS7, each protein has an SH2 domain and a 40-amino-acid region called the SOCS box.[53] The SOCS box can interact with a number of proteins to form a protein complex, and this complex can then cause the breakdown of JAKs and the receptors themselves, therefore inhibiting JAK-STAT signalling.[4] The protein complex does this by allowing a marker called ubiquitin to be added to proteins, in a process called ubiquitination, which signals for a protein to be broken down.[54] The proteins, such as JAKs and the receptors, are then transported to a compartment in the cell called the proteasome, which carries out protein breakdown.[54]
SOCS can also function by binding to proteins involved in JAK-STAT signalling and blocking their activity. For example, the SH2 domain of SOCS1 binds to a tyrosine in the activation loop of JAKs, which prevents JAKs from phosphorylating each other.[4] The SH2 domains of SOCS2, SOCS3 and CIS bind directly to receptors themselves.[54] Also, SOCS1 and SOCS3 can prevent JAK-STAT signalling by binding to JAKs, using segments called kinase inhibitory regions (KIRs) and stopping JAKs binding to other proteins.[55] The exact details of how other SOCS function is less understood.[4]
| Regulator | Positive or Negative regulation | Function |
|---|---|---|
| PTPs | SHP-1 and SHP-2: Negative, but could also be positive. CD45, PTP1B, TC-PTP: Negative | Removes phosphate groups from receptors, JAKs and STATs |
| SOCS | Negative | SOCS1 and SOCS3 block JAKs active sites using KIR domains. SOCS2, SOCS3 and CIS can bind receptors. SOCS1 and SOCS3 can signal JAKs and receptor for degradation. |
| PIAS | Negative | Add SUMO group to STATs to inhibit STAT activity. Recruit histone deacetylases to lower gene expression. Prevent STATs binding to DNA. |
Clinical significance
Since the JAK-STAT pathway plays a major role in many fundamental processes, such as apoptosis and inflammation, dysfunctional proteins in the pathway may lead to a number of diseases. For example, alterations in JAK-STAT signalling can result in cancer and diseases affecting the immune system, such as severe combined immunodeficiency disorder (SCID).[56]
Immune system-related diseases
JAK3 can be used for the signalling of IL-2, IL-4, IL-15 and IL-21 (as well as other cytokines); therefore patients with mutations in the JAK3 gene often experience issues affecting many aspects of the immune system.[57][58] For example, non-functional JAK3 causes SCID, which results in patients having no NK cells, B cells or T cells, and this would make SCID individuals susceptible to infection.[58] Mutations of the STAT5 protein, which can signal with JAK3, has been shown to result in autoimmune disorders.[59]
It has been suggested that patients with mutations in STAT1 and STAT2 are often more likely to develop infections from bacteria and viruses.[60] Also, STAT4 mutations have been associated with rheumatoid arthritis, and STAT6 mutations are linked to asthma.[61][62]
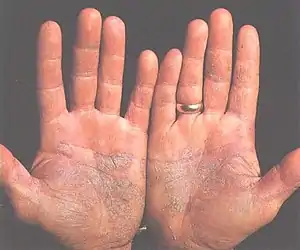
Patients with a faulty JAK-STAT signalling pathway may also experience skin disorders. For example, non-functional cytokine receptors, and overexpression of STAT3 have both been associated with psoriasis (an autoimmune disease associated with red, flaky skin).[58] STAT3 plays an important role in psoriasis, as STAT3 can control the production of IL-23 receptors, and IL-23 can help the development of Th17 cells, and Th17 cells can induce psoriasis.[63] Also, since many cytokines function through the STAT3 transcription factor, STAT3 plays a significant role in maintaining skin immunity.[58] In addition, because patients with JAK3 gene mutations have no functional T cells, B cells or NK cells, they would more likely to develop skin infections.
Cancer
Cancer involves abnormal and uncontrollable cell growth in a part of the body. Therefore, since JAK-STAT signalling can allow the transcription of genes involved in cell division, one potential effect of excessive JAK-STAT signalling is cancer formation. High levels of STAT activation have been associated with cancer; in particular, high amounts of STAT3 and STAT5 activation is mostly linked to more dangerous tumours.[64] For example, too much STAT3 activity has been associated with increasing the likelihood of melanoma (skin cancer) returning after treatment and abnormally high levels of STAT5 activity have been linked to a greater probability of patient death from prostate cancer.[65][64] Altered JAK-STAT signalling can also be involved in developing breast cancer. JAK-STAT signalling in mammary glands (located within breasts) can promote cell division and reduce cell apoptosis during pregnancy and puberty, and therefore if excessively activated, cancer can form.[66] High STAT3 activity plays a major role in this process, as it can allow the transcription of genes such as BCL2 and c-Myc, which are involved in cell division.[66]
Mutations in JAK2 can lead to leukaemia and lymphoma.[6] Specifically, mutations in exons 12, 13, 14 and 15 of the JAK2 gene are proposed to be a risk factor in developing lymphoma or leukemia.[6] Additionally, mutated STAT3 and STAT5 can increase JAK-STAT signalling in NK and T cells, which promotes very high proliferation of these cells, and increases the likelihood of developing leukaemia.[66] Also, a JAK-STAT signalling pathway mediated by erythropoietin (EPO), which usually allows the development of red blood cells, may be altered in patients with leukemia.[67]
Treatments
Since excessive JAK-STAT signalling is responsible for some cancers and immune disorders, JAK inhibitors have been proposed as drugs for therapy. For instance, to treat some forms of leukaemia, targeting and inhibiting JAKs could eliminate the effects of EPO signalling and perhaps prevent the development of leukaemia.[67] One example of a JAK inhibitor drug is Ruxolitinib, which is used as a JAK2 inhibitor.[64] STAT inhibitors are also being developed, and many of the inhibitors target STAT3.[66] It has been reported that therapies which target STAT3 can improve the survival of patients with cancer.[66] Another drug, called Tofacitinib, has been used for psoriasis and rheumatoid arthritis treatment, and has been approved for treatment of Crohn's disease and ulcerative colitis.[56]
See also
- Janus kinase inhibitor, a type of Janus kinases-blocking drugs used for cancer therapy.
- Signal transducing adaptor protein, a helper protein used by major proteins in signalling pathways.
References
- Aaronson DS, Horvath CM (2002). "A road map for those who don't know JAK-STAT". Science. 296 (5573): 1653–5. Bibcode:2002Sci...296.1653A. doi:10.1126/science.1071545. PMID 12040185. S2CID 20857536.
- Schindler, Christian; Levy, David E.; Decker, Thomas (2007). "JAK-STAT Signaling: From Interferons to Cytokines". Journal of Biological Chemistry. 282 (28): 20059–20063. doi:10.1074/jbc.R700016200. PMID 17502367.
- Kaneko, Tomonori; Joshi, Rakesh; Feller, Stephan M; Li, Shawn SC (2012). "Phosphotyrosine recognition domains: the typical, the atypical and the versatile". Cell Communication and Signaling. 10 (1): 32. doi:10.1186/1478-811X-10-32. PMC 3507883. PMID 23134684.
- Kiu, Hiu; Nicholson, Sandra E. (2012). "Biology and significance of the JAK/STAT signalling pathways". Growth Factors. 30 (2): 88–106. doi:10.3109/08977194.2012.660936. PMC 3762697. PMID 22339650.
- Kisseleva; Bhattacharya, S; Braunstein, J; Schindler, CW; et al. (2002-02-20). "Signaling through the JAK/STAT pathway, recent advances and future challenges". Gene. 285 (1–2): 1–24. doi:10.1016/S0378-1119(02)00398-0. PMID 12039028. November 2020
- Jatiani, S. S.; Baker, S. J.; Silverman, L. R.; Reddy, E. P. (2011). "JAK/STAT Pathways in Cytokine Signaling and Myeloproliferative Disorders: Approaches for Targeted Therapies". Genes & Cancer. 1 (10): 979–993. doi:10.1177/1947601910397187. PMC 3063998. PMID 21442038.
- Freund, P.; Kerenyi, M. A.; Hager, M.; Wagner, T.; Wingelhofer, B.; Pham, H T T.; Elabd, M.; Han, X.; Valent, P.; Gouilleux, F.; Sexl, V.; Krämer, O. H.; Groner, B.; Moriggl, R. (2017). "O-GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies". Leukemia. 31 (10): 2132–2142. doi:10.1038/leu.2017.4. PMC 5629373. PMID 28074064.
- Reich, Nancy C; Rout, M. P. (2014). "STATs get their move on". JAK-STAT. 2 (4): 27080. doi:10.4161/jkst.27080. PMC 3891633. PMID 24470978.
- Liu, L.; McBride, K. M.; Reich, N. C. (2005). "STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin- 3". Proceedings of the National Academy of Sciences. 102 (23): 8150–8155. doi:10.1073/pnas.0501643102. PMC 1149424. PMID 15919823.
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M. R.; Chen, X.; Du, Y.; Wang, Y.; An, L.; Wang, Q.; Lu, T.; Zhang, X.; Wang, Z.; Stark, G. R. (2010). "Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes". Proceedings of the National Academy of Sciences. 107 (50): 21499–21504. Bibcode:2010PNAS..10721499Y. doi:10.1073/pnas.1016147107. PMC 3003019. PMID 21098664.
- Stark, George R.; Darnell, James E. (2012). "The JAK-STAT Pathway at Twenty". Immunity. 36 (4): 503–514. doi:10.1016/j.immuni.2012.03.013. PMC 3909993. PMID 22520844.
- Zhuang, Shougang (2013). "Regulation of STAT signaling by acetylation". Cellular Signalling. 25 (9): 1924–1931. doi:10.1016/j.cellsig.2013.05.007. PMC 4550442. PMID 23707527.
- Ma, L.; Gao, J.-s.; Guan, Y.; Shi, X.; Zhang, H.; Ayrapetov, M. K.; Zhang, Z.; Xu, L.; Hyun, Y.-M.; Kim, M.; Zhuang, S.; Chin, Y. E. (2010). "Acetylation modulates prolactin receptor dimerization". Proceedings of the National Academy of Sciences. 107 (45): 19314–19319. Bibcode:2010PNAS..10719314M. doi:10.1073/pnas.1010253107. PMC 2984224. PMID 20962278.
- Shen, Y.; Schlessinger, K.; Zhu, X.; Meffre, E.; Quimby, F.; Levy, D. E.; Darnell, J. E. (2003). "Essential Role of STAT3 in Postnatal Survival and Growth Revealed by Mice Lacking STAT3 Serine 727 Phosphorylation". Molecular and Cellular Biology. 24 (1): 407–419. doi:10.1128/MCB.24.1.407-419.2004. PMC 303338. PMID 14673173.
- Decker, Thomas; Kovarik, Pavel (2000). "Serine phosphorylation of STATs". Oncogene. 19 (21): 2628–2637. doi:10.1038/sj.onc.1203481. PMID 10851062.
- Paulson, Matthew; Press, Carolyn; Smith, Eric; Tanese, Naoko; Levy, David E. (2002). "IFN-Stimulated transcription through a TBP-free acetyltransferase complex escapes viral shutoff". Nature Cell Biology. 4 (2): 140–147. doi:10.1038/ncb747. PMID 11802163. S2CID 20623715.
- Rawlings, Jason S.; Rosler, Kristin M.; Harrison, Douglas A. (2004). "The JAK/STAT signaling pathway". Journal of Cell Science. 117 (8): 1281–1283. doi:10.1242/jcs.00963. PMID 15020666.
- Jain, Neeraj; Zhang, Tong; Fong, Siok Lyn; Lim, Cheh Peng; Cao, Xinmin (1998). "Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK)". Oncogene. 17 (24): 3157–3167. doi:10.1038/sj.onc.1202238. PMID 9872331.
- Malek, Thomas R.; Castro, Iris (2010). "Interleukin-2 Receptor Signaling: At the Interface between Tolerance and Immunity". Immunity. 33 (2): 153–165. doi:10.1016/j.immuni.2010.08.004. PMC 2946796. PMID 20732639.
- Sen, B.; Saigal, B.; Parikh, N.; Gallick, G.; Johnson, F. M. (2009). "Sustained Src Inhibition Results in Signal Transducer and Activator of Transcription 3 (STAT3) Activation and Cancer Cell Survival via Altered Janus-Activated Kinase-STAT3 Binding". Cancer Research. 69 (5): 1958–1965. doi:10.1158/0008-5472.CAN-08-2944. PMC 2929826. PMID 19223541.
- Smith, Geoffrey A; Uchida, Kenji; Weiss, Arthur; Taunton, Jack (2016). "Essential biphasic role for JAK3 catalytic activity in IL-2 receptor signaling". Nature Chemical Biology. 12 (5): 373–379. doi:10.1038/nchembio.2056. PMC 4837022. PMID 27018889.
- Rodig, Scott J; Meraz, Marco A; White, J.Michael; Lampe, Pat A; Riley, Joan K; Arthur, Cora D; King, Kathleen L; Sheehan, Kathleen C.F; Yin, Li; Pennica, Diane; Johnson, Eugene M; Schreiber, Robert D (1998). "Disruption of the Jak1 Gene Demonstrates Obligatory and Nonredundant Roles of the Jaks in Cytokine-Induced Biologic Responses". Cell. 93 (3): 373–383. doi:10.1016/S0092-8674(00)81166-6. PMID 9590172. S2CID 18684846.
- Grebien, F.; Kerenyi, M. A.; Kovacic, B.; Kolbe, T.; Becker, V.; Dolznig, H.; Pfeffer, K.; Klingmuller, U.; Muller, M.; Beug, H.; Mullner, E. W.; Moriggl, R. (2008). "Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2". Blood. 111 (9): 4511–4522. doi:10.1182/blood-2007-07-102848. PMC 2976848. PMID 18239084.
- Luo, Hong; Dearolf, Charles R. (2001). "The JAK/STAT pathway andDrosophila development". BioEssays. 23 (12): 1138–1147. doi:10.1002/bies.10016. PMID 11746233. S2CID 41826277.
- Luo, H; Rose, P; Barber, D; Hanratty, W P; Lee, S; Roberts, T M; D'Andrea, A D; Dearolf, C R (1997). "Mutation in the Jak kinase JH2 domain hyperactivates Drosophila and mammalian JAK-STAT pathways". Molecular and Cellular Biology. 17 (3): 1562–1571. doi:10.1128/MCB.17.3.1562. PMC 231882. PMID 9032284.
- Binari, R; Perrimon, N (1994). "Stripe-specific regulation of pair-rule genes by hopscotch, a putative Jak family tyrosine kinase in Drosophila". Genes & Development. 8 (3): 300–312. doi:10.1101/gad.8.3.300. PMID 8314084.
- Yan, Riqiang; Small, Stephen; Desplan, Claude; Dearolf, Charles R; Darnell, James E; Roberts, T M; D'Andrea, A D; Dearolf, C R (1996). "Identification of a Stat Gene That Functions in Drosophila Development". Cell. 84 (3): 421–430. doi:10.1016/S0092-8674(00)81287-8. PMID 8608596. S2CID 15765894.
- Shuai K (2006). "Regulation of cytokine signaling pathways by PIAS proteins". Cell Research. 16 (2): 196–202. doi:10.1038/sj.cr.7310027. PMID 16474434. 16474434.
- Henenstreit, D.; Horeks-Hoeck, J.; Duschl, A. (2005). "JAK/STAT-dependent gene regulation by cytokines". Drug News & Perspectives. 18 (4): 243–9. doi:10.1358/dnp.2005.18.4.908658. PMID 16034480.
- Krebs DL, Hilton DJ (2001). "SOCS proteins: negative regulators of cytokine signaling". Stem Cells. 19 (5): 378–87. doi:10.1634/stemcells.19-5-378. PMID 11553846. S2CID 20847942.
- Yamada, Satoshi; Shiono, Satoru; Joo, Akiko; Yoshimura, Akihiko (23 December 2002). "Control mechanism of JAK/STAT signal transduction pathway". FEBS Letters. 534 (1–3): 190–196. doi:10.1016/s0014-5793(02)03842-5. ISSN 0014-5793. PMID 12527385. S2CID 38090088.
- Singh, Abhay; Jayaraman, Arul; Hahn, Juergen (2006). "Modeling regulatory mechanisms in IL-6 signal transduction in hepatocytes". Biotechnology and Bioengineering. 95 (5): 850–862. doi:10.1002/bit.21026. ISSN 1097-0290. PMID 16752369. S2CID 20924311.
- Mortlock, Ryland D.; Georgia, Senta K.; Finley, Stacey D. (1 February 2021). "Dynamic Regulation of JAK-STAT Signaling Through the Prolactin Receptor Predicted by Computational Modeling". Cellular and Molecular Bioengineering. 14 (1): 15–30. doi:10.1007/s12195-020-00647-8. ISSN 1865-5033. PMC 7878662. PMID 33633812.
- Shuai, Ke; Liu, Bin; Zhang, Di; Cui, Yan; Zhou, Jinlian; Cui, Sheng (2005). "Regulation of gene-activation pathways by PIAS proteins in the immune system". Nature Reviews Immunology. 5 (8): 593–605. doi:10.1038/nri1667. PMID 16056253. S2CID 7466028.
- Ungureanu, D.; Vanhatupa, S.; Grönholm, J.; Palvimo, J.; Silvennoinen, O. (2005). "SUMO-1 conjugation selectively modulates STAT1-mediated gene responses". Blood. 106 (1): 224–226. doi:10.1182/blood-2004-11-4514. PMID 15761017.
- Droescher, Mathias; Begitt, Andreas; Marg, Andreas; Zacharias, Martin; Vinkemeier, Uwe (2011). "Cytokine-induced Paracrystals Prolong the Activity of Signal Transducers and Activators of Transcription (STAT) and Provide a Model for the Regulation of Protein Solubility by Small Ubiquitin-like Modifier (SUMO)". Journal of Biological Chemistry. 286 (21): 18731–18746. doi:10.1074/jbc.M111.235978. PMC 3099690. PMID 21460228.
- Liu, B.; Gross, M.; ten Hoeve, J.; Shuai, K. (2001). "A transcriptional corepressor of Stat1 with an essential LXXLL signature motif". Proceedings of the National Academy of Sciences. 98 (6): 3203–3207. Bibcode:2001PNAS...98.3203L. doi:10.1073/pnas.051489598. PMC 30631. PMID 11248056.
- Xu, Dan; Qu, Cheng-Kui (2008). "Protein tyrosine phosphatases in the JAK/STAT pathway". Frontiers in Bioscience. 13 (1): 4925–4932. doi:10.2741/3051. PMC 2599796. PMID 18508557.
- Yi, T L; Cleveland, J L; Ihle, J N (1992). "Protein tyrosine phosphatase containing SH2 domains: characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12-p13". Molecular and Cellular Biology. 12 (2): 836–846. doi:10.1128/MCB.12.2.836. PMC 364317. PMID 1732748.
- M. Scott, Latanya; R. Lawrence, Harshani; M. Sebti, Said; J. Lawrence, Nicholas; Wu, Jie (2010). "Targeting Protein Tyrosine Phosphatases for Anticancer Drug Discovery". Current Pharmaceutical Design. 16 (16): 1843–1862. doi:10.2174/138161210791209027. PMC 3076191. PMID 20337577.
- Bone, Heather; Dechert, Ute; Jirik, Frank; Schrader, John W.; Welham, Melanie J. (1997). "SHP1 and SHP2 Protein-tyrosine Phosphatases Associate with βc after Interleukin-3-induced Receptor Tyrosine Phosphorylation". Journal of Biological Chemistry. 272 (22): 14470–14476. doi:10.1074/jbc.272.22.14470. PMID 9162089.
- Lyons, Bonnie L; Lynes, Michael A; Burzenski, Lisa; Joliat, Melissa J; Hadjout, Nacima; Shultz, Leonard D (2003). "Mechanisms of anemia in SHP-1 protein tyrosine phosphatase-deficient "viable motheaten" mice". Experimental Hematology. 31 (3): 234–243. doi:10.1016/S0301-472X(02)01031-7. PMID 12644021.
- Johan, M. F.; Bowen, D. T.; Frew, M. E.; Goodeve, A. C.; Reilly, J. T. (2005). "Aberrant methylation of the negative regulators RASSFIA, SHP-1 and SOCS-1 in myelodysplastic syndromes and acute myeloid leukaemia". British Journal of Haematology. 129 (1): 60–65. doi:10.1111/j.1365-2141.2005.05412.x. PMID 15801956. S2CID 25021813.
- You, Min; Zhao, Zhizhuang (1997). "Positive Effects of SH2 Domain-containing Tyrosine Phosphatase SHP-1 on Epidermal Growth Factor- and Interferon-γ-stimulated Activation of STAT Transcription Factors in HeLa Cells". Journal of Biological Chemistry. 272 (37): 23376–23381. doi:10.1074/jbc.272.37.23376. PMID 9287352.
- Neel, Benjamin G.; Gu, Haihua; Pao, Lily (2003). "The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling". Trends in Biochemical Sciences. 28 (6): 284–293. doi:10.1016/S0968-0004(03)00091-4. PMID 12826400.
- Wu, Tong R.; Hong, Y. Kate; Wang, Xu-Dong; Ling, Mike Y.; Dragoi, Ana M.; Chung, Alicia S.; Campbell, Andrew G.; Han, Zhi-Yong; Feng, Gen-Sheng; Chin, Y. Eugene (2002). "SHP-2 Is a Dual-specificity Phosphatase Involved in Stat1 Dephosphorylation at Both Tyrosine and Serine Residues in Nuclei". Journal of Biological Chemistry. 277 (49): 47572–47580. doi:10.1074/jbc.M207536200. PMID 12270932.
- Chen, Yuhong; Wen, Renren; Yang, Shoua; Schuman, James; Zhang, Eric E.; Yi, Taolin; Feng, Gen-Sheng; Wang, Demin (2003). "Identification of Shp-2 as a Stat5A Phosphatase". Journal of Biological Chemistry. 278 (19): 16520–16527. doi:10.1074/jbc.M210572200. PMID 12615921.
- Zhang, E. E.; Chapeau, E.; Hagihara, K.; Feng, G.-S. (2004). "Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism". Proceedings of the National Academy of Sciences. 101 (45): 16064–16069. Bibcode:2004PNAS..10116064Z. doi:10.1073/pnas.0405041101. PMC 528739. PMID 15520383.
- Ke, Yuehai; Lesperance, Jacqueline; Zhang, Eric E.; Bard-Chapeau, Emilie A.; Oshima, Robert G.; Muller, William J.; Feng, Gen-Sheng (2006). "Conditional Deletion of Shp2 in the Mammary Gland Leads to Impaired Lobulo-alveolar Outgrowth and Attenuated Stat5 Activation". Journal of Biological Chemistry. 281 (45): 34374–34380. doi:10.1074/jbc.M607325200. PMC 1761121. PMID 16959766.
- Yu, Wen-Mei; Hawley, Teresa S; Hawley, Robert G; Qu, Cheng-Kui (2003). "Catalytic-dependent and -independent roles of SHP-2 tyrosine phosphatase in interleukin-3 signaling". Oncogene. 22 (38): 5995–6004. doi:10.1038/sj.onc.1206846. PMID 12955078.
- Yamada, Takechiyo; Zhu, Daocheng; Saxon, Andrew; Zhang, Ke (2002). "CD45 Controls Interleukin-4-mediated IgE Class Switch Recombination in Human B Cells through Its Function as a Janus Kinase Phosphatase". Journal of Biological Chemistry. 277 (32): 28830–28835. doi:10.1074/jbc.M201781200. PMID 11994288.
- Irie-Sasaki, Junko; Sasaki, Takehiko; Matsumoto, Wataru; Opavsky, Anne; Cheng, Mary; Welstead, Grant; Griffiths, Emily; Krawczyk, Connie; Richardson, Christopher D.; Aitken, Karen; Iscove, Norman; Koretzky, Gary; Johnson, Pauline; Liu, Peter; Rothstein, David M.; Penninger, Josef M. (2001). "CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling". Nature. 409 (6818): 349–354. Bibcode:2001Natur.409..349I. doi:10.1038/35053086. PMID 11201744. S2CID 4423377.
- Alexander, Warren S.; Hilton, Douglas J. (2004). "The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response". Annual Review of Immunology. 22 (1): 503–529. doi:10.1146/annurev.immunol.22.091003.090312. PMID 15032587.
- Tamiya, T.; Kashiwagi, I.; Takahashi, R.; Yasukawa, H.; Yoshimura, A. (2011). "Suppressors of Cytokine Signaling (SOCS) Proteins and JAK/STAT Pathways: Regulation of T-Cell Inflammation by SOCS1 and SOCS3". Arteriosclerosis, Thrombosis, and Vascular Biology. 31 (5): 980–985. doi:10.1161/ATVBAHA.110.207464. PMID 21508344.
- Kershaw, Nadia J.; Murphy, James M.; Lucet, Isabelle S.; Nicola, Nicos A.; Babon, Jeffrey J. (2013). "Regulation of Janus kinases by SOCS proteins". Biochemical Society Transactions. 41 (4): 1042–1047. doi:10.1042/BST20130077. PMC 3773493. PMID 23863176.
- Villarino, Alejandro V.; Kanno, Yuka; Ferdinand, John R.; O’Shea, John J. (2015). "Mechanisms of Jak/STAT Signaling in Immunity and Disease". The Journal of Immunology. 194 (1): 21–27. doi:10.4049/jimmunol.1401867. PMC 4524500. PMID 25527793.
- Pesu, Marko; Candotti, Fabio; Husa, Matthew; Hofmann, Sigrun R.; Notarangelo, Luigi D.; O'Shea, John J. (2005). "Jak3, severe combined immunodeficiency, and a new class of immunosuppressive drugs". Immunological Reviews. 203 (1): 127–142. doi:10.1111/j.0105-2896.2005.00220.x. PMID 15661026. S2CID 20684919.
- Welsch, Katharina; Holstein, Julia; Laurence, Arian; Ghoreschi, Kamran (2017). "Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors". European Journal of Immunology. 47 (7): 1096–1107. doi:10.1002/eji.201646680. PMID 28555727.
- Casanova, Jean-Laurent; Holland, Steven M.; Notarangelo, Luigi D. (2012). "Inborn Errors of Human JAKs and STATs". Immunity. 36 (4): 515–528. doi:10.1016/j.immuni.2012.03.016. PMC 3334867. PMID 22520845.
- Au-Yeung, Nancy; Mandhana, Roli; Horvath, Curt M (2014). "Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway". JAK-STAT. 2 (3): 23931. doi:10.4161/jkst.23931. PMC 3772101. PMID 24069549.
- Remmers, Elaine F.; Plenge, Robert M.; Lee, Annette T.; Graham, Robert R.; Hom, Geoffrey; Behrens, Timothy W.; de Bakker, Paul I.W.; Le, Julie M.; Lee, Hye-Soon; Batliwalla, Franak; Li, Wentian; Masters, Seth L.; Booty, Matthew G.; Carulli, John P.; Padyukov, Leonid; Alfredsson, Lars; Klareskog, Lars; Chen, Wei V.; Amos, Christopher I.; Criswell, Lindsey A.; Seldin, Michael F.; Kastner, Daniel L.; Gregersen, Peter K. (2007). "STAT4 and the Risk of Rheumatoid Arthritis and Systemic Lupus Erythematosus". New England Journal of Medicine. 357 (10): 977–986. doi:10.1056/NEJMoa073003. PMC 2630215. PMID 17804842.
- Vercelli, Donata (2008). "Discovering susceptibility genes for asthma and allergy". Nature Reviews Immunology. 8 (3): 169–182. doi:10.1038/nri2257. PMID 18301422. S2CID 27558099.
- Ghoreschi, Kamran; Laurence, Arian; Yang, Xiang-Ping; Hirahara, Kiyoshi; O'Shea, John J. (2011). "T helper 17 cell heterogeneity and pathogenicity in autoimmune disease". Trends in Immunology. 32 (9): 395–401. doi:10.1016/j.it.2011.06.007. PMC 3163735. PMID 21782512.
- Thomas, S J; Snowden, J A; Zeidler, M P; Danson, S J (2015). "The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours". British Journal of Cancer. 113 (3): 365–371. doi:10.1038/bjc.2015.233. PMC 4522639. PMID 26151455.
- Messina, Jane L.; Yu, Hua; Riker, Adam I.; Munster, Pamela N.; Jove, Richard L.; Daud, Adil I. (2008). "Activated Stat-3 in Melanoma". Cancer Control. 15 (3): 196–201. doi:10.1177/107327480801500302. PMID 18596671.
- Groner, Bernd; von Manstein, Viktoria (2017). "Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition". Molecular and Cellular Endocrinology. 451: 1–14. doi:10.1016/j.mce.2017.05.033. PMID 28576744. S2CID 3833538.
- Kim, Jinkoo; Jung, Younghun; Sun, Hongli; Joseph, Jeena; Mishra, Anjali; Shiozawa, Yusuke; Wang, Jingcheng; Krebsbach, Paul H.; Taichman, Russell S. (2012). "Erythropoietin mediated bone formation is regulated by mTOR signaling". Journal of Cellular Biochemistry. 113 (1): 220–228. doi:10.1002/jcb.23347. PMC 3237787. PMID 21898543.
Further reading
- Schroder K, Hertzog PJ, Ravasi T, Hume DA (February 2004). "Interferon-gamma: an overview of signals, mechanisms and functions". Journal of Leukocyte Biology. 75 (2): 163–89. doi:10.1189/jlb.0603252. PMID 14525967.
- O'Shea JJ, Gadina M, Schreiber RD (April 2002). "Cytokine signaling in 2002: new surprises in the Jak/Stat pathway". Cell. 109 Suppl (Suppl): S121-31. doi:10.1016/S0092-8674(02)00701-8. PMID 11983158. S2CID 8251837.