Phakomatosis
Phakomatoses, also known neurocutaneous syndromes, are a group of multisystemic diseases that most prominently affect structures primarily derived from the ectoderm such as the central nervous system, skin and eyes. The majority of phakomatoses are single-gene disorders that may be inherited in an autosomal dominant, autosomal recessive or X-linked pattern. Presentations may vary dramatically between patients with the same particular syndrome due to mosaicism, variable expressivity, and penetrance.[1]
| Phakomatoses | |
|---|---|
| Other names | Neurocutaneous syndromes |
| Specialty | Medicine, Neurology, Neurosurgery, Medical Genetics, Dermatology, Psychology, Psychiatry and more |
| Symptoms | Dermal, ocular and CNS benign and malignant tumors. Various additional potential complications. |
| Complications | Numerous potential complications including cosmetic, intellectual disability, epilepsy, organ failure and more. |
| Usual onset | Childhood (most commonly) |
| Duration | Lifelong |
| Causes | Genetic causes |
| Treatment | Highly variable. Many require lifelong surveillance and various treatments depending on the particular syndrome and presentation. |
Many phakomatoses are caused by mutations which alter functioning of the RAS–mitogen-activated protein kinase (MAPK) pathway that regulates cellular growth, differentiation, proliferation and death.[2] This results in a tendency for individuals with these mutations to develop various types of benign or malignant tumors depending on the particular mutation. The presence of these tumors may result in functional and/or cosmetic problems depending on their type and location.
History
The term phakomatosis originated in 1923, when the Dutch ophthalmologist van der Hoeve[3] used the term phakoma to refer to a "mother spot" or birthmark,[3] a physical characteristic common to patients with tuberous sclerosis and neurofibromatosis that he examined.[3] The term phakomatoses was derived from phakos,[4] the Greek term for 'birthmark'.[4] He originally used the phrase to describe two diseases: neurofibromatosis and tuberous sclerosis.[4] This term later became imprecise when van der Hoeve also used it to include those with Sturge-Weber syndrome as they do not have similar lesions on the skin. In addition, the term phakomatosis makes no reference to the central nervous system involvement. The term neurocutaneous syndrome was subsequently defined by the Russian-American neurologist Paul Ivan Yakovlev and psychiatrist Riley H. Guthrie and it is currently more commonly used.[2]
The first clinical description was made in “Monstrorum Historia” by an Italian physician and naturalist named Ulisse Aldrovandi who described a patient with probable neurofibromatosis type I in 1592. He described a short man with a large tumor which was likely a plexiform neurofibroma.[2] However, there are artistic or descriptive representations which have been speculated to depict individuals with phakomatoses as far back as the Hellenistic period and ancient Egypt.[5][6]
Over time, the number of neurocutaneous syndromes have increased and there are several dozen that have been characterized.
Types
There are a large number of neurocutaneous syndromes that exceed the scope of this article. Therefore, characteristics of a few of the more common types are summarized.
Neurofibromatosis Type I (von Recklinghausen disease)

Neurofibromatosis type 1 is the most common phakomatosis and it affects approximately 1 in 2500-3000 live births.[7] It is a genetic disorder due to a germline mutation in the NF1 gene. This gene encodes a protein called neurofibromin that is involved in controlling cellular growth.[8] Malfunction of the gene results in multisystem manifestations involving the skin, central nervous system, peripheral nervous system, eyes and musculoskeletal system. The condition is inherited in an autosomal dominant manner. However, approximately one-half of patients with this condition have no family history and the mutation occurs spontaneously. In most instances, neurofibromatosis type 1 can be diagnosed clinically according to consensus criteria and genetic testing is only used in atypical presentations or for family planning decisions.[9]
Café au lait spots are one of the most characteristic features of neurofibromatosis type 1. They are hyperpigmented lesions of the skin that increase in number and size during the first years of life. They are present in almost all patients but do not have malignant potential.[10][11] Freckling in the axillary and inguinal regions are another common presenting feature seen in as many as 90% of patients during childhood.[10] Lisch nodules (benign hamartomas of the iris) are seen in almost all patients but they do not cause any visual or ocular impairment.
Neurofibromas are benign nerve sheath tumors that occur in peripheral nerves. These typically develop during the teenage years. Neurofibromas do not become malignant but can cause cosmetic concerns as well as local pruritus. When present in the spine they can affect nerve roots and result in both motor and sensory defects.[11] Surgery may be required in some cases. On the contrary, plexiform neurofibromas arise from multiple nerve fascicles and malignant transformation occurs in approximately 10% of cases.[12] They may result in significant morbidity as they may cause organ compression, vascular occlusions, bone destruction, pain and cosmetic issues. Plexiform neurofibromas are seen in 30-50% of patients.[10]
Optic pathway gliomas are seen in 15-20% of patients with neurofibromatosis type 1.[10] They most often arise during childhood. They often do not cause additional morbidity but may in up to one-half of patients.[13] Neurocognitive impairment, attention-deficit hyperactivity disorder, autism spectrum disorder and behavioral disorders are also commonly seen in patients with neurofibromatosis type 1.[14] Epilepsy is seen in 4-7% of patients.[15]
Musculoskeletal system manifestations can develop in patients with neurofibromatosis type 1. Common findings include sphenoid wing dysplasia, osteopenia, osteoporosis, anterior chest wall deformities as well as scoliosis. Patients are at a much greater risk for fractures than the general population.[11]
Vascular abnormalities are also frequently encountered in patients with neurofibromatosis type 1. These may include renal artery stenosis, pulmonary artery stenosis, cerebral artery stenosis and aneurysms.[11] Complications may include myocardial infarction and stroke.[16]
Relative to the general population the risk for certain types of cancer is increased significantly. For instance, the risk for brain tumors and breast cancer is increased by 5 times.[11]
Neurofibromatosis Type II

Neurofibromatosis type 2 is an autosomal dominant condition that affects approximately 1 in 35,000-40,000 people.[18] It is caused by mutations in the NF2 gene on chromosome 22 which has a high penetrance though most patients do not present with symptoms until adulthood.[19] Approximately half of patients have de novo mutations and as many as 59.7% are mosaic.[20][21] Patients who present in childhood tend to have a more severe phenotype.[22]
Bilateral vestibular schwannomas are the most characteristic finding and in the vast majority of cases are benign. However, they are responsible for significant morbidity and are responsible for the most common presenting symptom of hearing loss in adults.[23] They can also less commonly present with dizziness or balance impairment. It is estimated that schwannomas occur in over 90% of patients.[24]
Meningiomas are the second most common tumor in NF2. Approximately half of patients have an intracranial meningioma and extramedullary spinal meningiomas occur in one-fifth of patients.[23] Relative to the general population, meningiomas in NF2 tend to occur at a younger age, are more likely to be multiple and are associated with increased mortality.[25]
Spinal cord ependymomas occur in 20-50% of patients but they are asymptomatic in the majority of cases.[23] In cases that they are symptomatic the specific presentation will depend upon where they are located. Potential symptoms include back pain, weakness and sensory changes.
Peripheral neuropathy eventually occurs in most patients with NF2. In some cases, a mononeuropathy affecting the facial nerve can occur as the first presenting symptom of NF2.[23] Other potential manifestations include focal amyotrophy, mononeuropathy multiplex or a severe generalized polyneuropathy in 3-5% of patients.[20]
Ophthalmologic manifestations are also common with 60-80% of patients developing cataracts.[23] Optic nerve meningiomas and retinal hamartomas can result in vision loss.
Approximately, 70% of patients will have cutaneous manifestations but only 10% have more than 10 lesions.[23] The majority of skin lesions are schwannomas but neurofibromas can also occur.
Tuberous sclerosis (Bourneville syndrome)

Tuberous sclerosis complex (TSC) is a multisystemic disorder due to autosomal dominant mutations in either TSC1 or TSC2 which results in the impaired inhibition of the mechanistic target of rapamycin (mTOR) signaling pathway.[26] This leads to impaired regulation of cellular proliferation, survival, homeostasis, migration and other critical functions.[27] The most typically affected organs include the brain, skin, kidney, heart and lung. The incidence of TSC is approximately 1 in 6,000 live births.[28] Similar to other neurocutaneous disorders there is variable penetrance and expressivity.[26] TSC1 mutations tend to have a less severe phenotype and are more likely to be familial.[26] A major development in the treatment of this condition occurred in 2010s when the FDA approved mTOR inhibitors for the treatment of several manifestations of TSC.
Epilepsy is among the most common manifestations of TSC and it occurs 80-90% of patients.[29] It usually presents during the first few years of life and is medically refractory in two-thirds of patients.[30] Approximately, one-third of patients have infantile spasms.[30] There are several types of brain lesions that can be found in TSC including subependymal nodules (SENs), cortical tubers and subependymal giant cell astrocytomas (SEGAs). SENs and cortical tubers occur in approximately 80% and 90% of patients, respectively.[31] SEGAs occur in 10-15% of patients and are a major potential cause of morbidity and potentially mortality.[32] They tend to present during the first 20 years of life. TSC-Associated Neuropsychiatric Disorder (TAND) refers to the behavioral, intellectual and psychiatric manifestations of TSC including autism spectrum disorder, attention deficit hyperactivity disorder, intellectual disability, depression and anxiety. Approximately 90% of patients will have at least 1 symptom of TAND.[33]
Lymphangioleiomyomatosis (LAM) occurs in the lung and may result in pneumothorax, cystic lung destruction and pleural effusions. Symptoms which occur as a result may include fatigue, chest pain and shortness of breath. It occurs in approximately 30-40% of women with TSC, up to 80% by the age of 40, and it is much less common in men.[34]
Renal angiomyolipomas and cysts are the most common manifestations of TSC involving the kidney. Renal disease is among the most common causes of early death in TSC. One study found that renal lesions were present in 80% of patients by a mean age of 10.5 years.[35] Renal cell carcinoma occurs in 2-5% of patients with TSC at a mean age of 28–30 years.[36]
Cardiac rhabdomyomas are benign hamartomas and are the most common cardiac manifestations of TSC. It is found in approximately two-third of newborns with TSC.[37] Most of the time they do not cause symptoms and spontaneously regress. In a minority of cases, they may result in heart failure, arrhythmias, and murmurs.[37]
Dermatological manifestations occur in almost all patients and may include facial angiofibromas, confetti skin lesions, ungual fibromas, shagreen patches, hypomelanotic macules and fibrous cephalic plaques.[13] None of these tend to result in significant complications however facial angiofibromas may cause significant cosmetic concerns.[38]
Sturge–Weber Syndrome
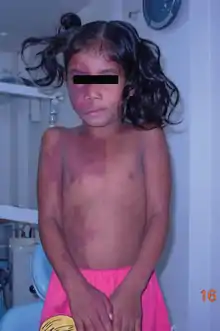
Sturge-Weber syndrome occurs in approximately 1 in 20,000-50,000 live births and is caused by a somatic activating mutation in GNAQ.[39][40] Normally, GNAQ is involved in cell growth signal transmission.[40] It is classically characterized by a facial port-wine stain in the ophthalmic division of the trigeminal nerve, glaucoma and leptomeningeal angioma.[39] However, the clinical presentation can vary significantly depending on the timing of the somatic mutation ranging from an isolated port-wine stain to complete Sturge-Weber syndrome.[39]
The characteristic port-wine stain, also called nevus flammeus, is caused by a capillary or venular malformation. It is present from birth while the extent and size of it is associated with the risk of leptomeningeal and ophthalmologic involvement.[41] The greatest risk is associated with port-wine stains that appear to involve the entire V1 distribution followed by partial V1 involvement.[41] There is ontroversy as to whether or not the distribution of port-wine stains truly follows trigeminal nerve branches per se.[42] Port-wine stains are most often unilateral but can be bilateral. Typically, brain and eye involvement occur on the same side as the port-wine stain. Over time, port-wine stains can develop soft tissue or bone hypertrophy, proliferative nodules, and progressive ectasia which can lead to significant disfigurement.[43]
The two most common ocular manifestations include glaucoma and choroidal hemagioma. Glaucoma is estimated to occur in 30-70% with a bimodal peak occurring at the time of birth in 60% and between childhood and adolescence in 40%.[44] The most frequent form of glaucoma is open-angle however closed-angle glaucoma may also occur.[44] Congenital onset glaucoma is associated with other changes of the eye including megalocornea and buphthalmos.[45] The choroidal hemangioma occurs in 40-50% of patients.[46] They are usually asymptomatic but they may become thickened overtime and may be associated with an increased risk for glaucoma.[44][47]
Seizures typically develop within the first two years of life and occur in 75-100% of patients.[48][39] Onset of seizures occur in 95% by the age of 5 years.[49] Status epilepticus occurs in approximately 50% of patients.[50] Approximately 25% of patients develop drug-resistant epilepsy.[51] In these cases, surgery may be pursued with the two main approaches being lesionectomy and hemispherectomy.[51]
Neurological impairment can accrue gradually over time and may occur in the context of stroke-like episodes which may be triggered by seizures or head injuries.[48] Intellectual disability occurs in approximately one-half of patients.[48] In addition, patients with Sturge-Weber syndrome have an increased prevalence of depression, endocrinological abnormalities, headaches with migraine-like features, ADHD and behavioral problems.[52] Cortically mediated visual field defects and hemiparesis occur in approximately one-third and one-half of patients, respectively.[48]
Von Hippel–Lindau syndrome (hemangiomatosis)
Von Hippel-Lindau (VHL) disease is an autosomal dominant condition caused by mutations of the VHL gene.[53] Approximately one-in-five cases are de novo rather than familial and it has nearly complete penetrance.[54] VHL occurs in an estimated 1 in 36,000-45,000 live births.[54] VHL normally functions as a tumor suppressor gene and thus when not functioning normally results in the development of benign and malignant tumors as well as cysts. Some of the most common manifestations include hemangioblastomas in the retina and central nervous system, clear cell renal cell carcinomas, pheochromocytomas, endolymphatic sac tumors and pancreatic neuroendocrine tumors. Life expectancy is reduced for individuals with this condition and one study found a median age of death at 52 years.[55]
CNS hemangioblastomas are the cardinal feature of VHL and occur in as many as 80% of cases.[56] The most common locations include the cerebellum, spinal cord and brainstem.[56] These tumors are benign however they may cause significant morbidity as well as mortality due to mass effect.[57] Tumor growth rates can be highly variable. Treatment options include resection and radiation.[57]
Retinal hemangioblastomas occur in approximately half of patients and are often the first presenting manifestation of VHL.[58] One longitudinal study that followed patients for a mean period of 7.3 years found that in individuals with unilateral disease 100% had bilateral involvement by the age of 56 years.[59] Blindness or severe visual impairment occurs in less than 10% of patients.[60][61] Laser photocoagulation and cryotherapy are the most common surgical treatments.[56]
Renal cell carcinoma is a major cause of death in patients with VHL and it occurs in 70% of patients.[56] Lesions larger than 3 cm are associated with a risk of metastasis and thus present a recommended threshold for resection.[62][63] Nephron sparing surgery allows for preservation of kidney function and 10-year survival rates up to 81%.[64]
Additional manifestations of VHL may include pheochromocytomas in as many as 16% of patients with VHL.[65] They are most often unilateral but can be bilateral or multifocal. Approximately 5% are malignant.[56]
Pancreatic involvement occurs in 77% of patients with VHL. Asymptomatic cysts consist of the majority of cases. Neuroendocrine tumors occur in approximately 15% of cases.[66] Less than 10% with neuroendocrine tumors will develop metastases.[67]
Genetics
The majority of neurocutaneous syndromes are single-gene disorders however they are caused by different genes and have different inheritance patterns. For instance, neurofibromatosis 1 and 2, Tuberous sclerosis complex, Von Hippel-Lindau syndrome and Legius syndrome are inherited in an autosomal dominant manner. Incontinentia pigmenti is X-linked dominant and Sturge-Weber syndrome is sporadic. Some neurocutaneous disorders are found exclusively as mosaics such as Sturge-Weber syndrome and Proteus syndrome. Others such as neurofibromatosis type 1 and 2 as well as tuberous sclerosis complex can potentially be mosaics but may not be.[68] Mosaicism may be suspected in cases with either a mild or incomplete presentation of a neurocutaneous disorder. A definitive diagnosis is most likely to be obtained if testing of affected tissues is possible.[69][70]
Patients and families of those affected by neurocutaneous disorders often benefit from genetic counseling. It may provide an opportunity to provide a better understanding of the potential risk to future offspring as well as to improve coping with the various implications of the condition. If desired, a prenatal diagnosis can be obtained by either amniocentesis or chorionic villus sampling. Another potential option is preimplantation genetic diagnosis where an embryo that does not have the mutation can be selectively implanted into the mother.[71]
Diagnosis
Many neurocutaneous syndromes have established diagnostic criteria which may facilitate a diagnosis without necessarily requiring genetic testing. For instance, neurofibromatosis types 1 and 2, tuberous sclerosis complex and incontinentia pigmenti have formal diagnostic criteria.[72] Whereas other syndromes such as Noonan syndrome with multiple lentigines, for instance, do not. In cases where conditions that do not have diagnostic criteria are suspected, genetic testing is more likely to be necessary. Diagnostic criteria are not perfect as sensitivity and specificity is not 100%.[73] Similarly, genetic testing can produce false negatives. For example, genetic testing is positive in only 75-90% of cases of tuberous sclerosis complex.[74] Thus, clinicians must apply clinical judgement when evaluating an individual suspected to have a neurocutaneous syndrome.
Treatment
The treatment of each neurocutaneous syndrome is unique. For some neurocutaneous syndromes such as neurofibromatosis 1 and tuberous sclerosis complex there are guidelines with recommendations for surveillance and management.[75][76] For less common syndromes such guidelines are not yet available. Surveillance is a necessity for many neurocutaneous syndromes because new manifestations may develop over time which may only be detected with specific and focused testing. Thus, patients may be advised to obtain particular evaluations (e.g., MRI, ophthalmologic or dermatological examinations) within recommended intervals over time with the aim of detecting new manifestations of the syndrome early on.
Most of the currently available treatments for neurocutaneous syndromes do not address the underlying genetic cause. For example, epilepsy surgery in tuberous sclerosis complex or vestibular schwannoma surgery in neurofibromatosis type 2. However, there are some treatments that do address the underlying cause such as the use of mTOR inhibitors in tuberous sclerosis complex. There are currently significant efforts underway to develop additional treatments that address the underlying causes of neurocutaneous syndromes.[77]
Neurocutaneous syndromes are complex, lifelong conditions that have the potential to affect many different organ systems over time. Thus, patients may benefit from a multidisciplinary approach for care. Integrated, multidisciplinary clinics have developed in an effort to optimize the long-term care for patients with neurocutaneous syndromes.[78] Specialties represented at these clinics may include genetics, neurology, ophthalmology, hematology-oncology, neurosurgery, psychiatry, dermatology and more.
References
- Gürsoy, Semra; Erçal, Derya (2018-10-10). "Genetic Evaluation of Common Neurocutaneous Syndromes". Pediatric Neurology. 89: 3–10. doi:10.1016/j.pediatrneurol.2018.08.006. ISSN 1873-5150. PMID 30424961. S2CID 53302650.
- Ruggieri, Martino; Praticò, Andrea D. (2015-12-17). "Mosaic Neurocutaneous Disorders and Their Causes". Seminars in Pediatric Neurology. 22 (4): 207–233. doi:10.1016/j.spen.2015.11.001. ISSN 1558-0776. PMID 26706010.
- Fernández-Guarino M, Boixeda P, de Las Heras E, Aboin S, García-Millán C, Olasolo PJ (January 2008). "Phakomatosis pigmentovascularis: Clinical findings in 15 patients and review of the literature". Journal of the American Academy of Dermatology. 58 (1): 88–93. doi:10.1016/j.jaad.2007.08.012. PMID 18045734.
- Huson SM, Korf BR (2013). "The Phakomatoses". Emery and Rimoin's Principles and Practice of Medical Genetics. Elsevier. pp. 1–45. doi:10.1016/b978-0-12-383834-6.00128-2. ISBN 978-0-12-383834-6.
- Ruggieri M, Gentile AE, Ferrara V, Papi M, Praticò AD, Mudry A, et al. (June 2021). "Neurocutaneous syndromes in art and antiquities". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 187 (2): 224–234. doi:10.1002/ajmg.c.31917. PMC 8252443. PMID 34013593.
- Ruggieri, Martino; Praticò, Andrea D.; Caltabiano, Rosario; Polizzi, Agata (2018-02-01). "Early history of the different forms of neurofibromatosis from ancient Egypt to the British Empire and beyond: First descriptions, medical curiosities, misconceptions, landmarks, and the persons behind the syndromes". American Journal of Medical Genetics Part A. 176 (3): 515–550. doi:10.1002/ajmg.a.38486. ISSN 1552-4825. PMID 29388340. S2CID 26042790.
- Lammert M, Friedman JM, Kluwe L, Mautner VF (January 2005). "Prevalence of neurofibromatosis 1 in German children at elementary school enrollment". Archives of Dermatology. 141 (1): 71–74. doi:10.1001/archderm.141.1.71. PMID 15655144.
- Gutmann DH, Parada LF, Silva AJ, Ratner N (October 2012). "Neurofibromatosis type 1: modeling CNS dysfunction". The Journal of Neuroscience. 32 (41): 14087–14093. doi:10.1523/jneurosci.3242-12.2012. PMC 3477849. PMID 23055477. S2CID 1747782.
- Tadini G, Brems H, Legius E (2020). "Proposal of New Diagnostic Criteria". Multidisciplinary Approach to Neurofibromatosis Type 1. Cham: Springer International Publishing. pp. 309–313. doi:10.1007/978-3-319-92450-2_21. ISBN 978-3-319-92449-6. S2CID 226576109. Retrieved 2022-07-14.
- Tabata MM, Li S, Knight P, Bakker A, Sarin KY (August 2020). "Phenotypic heterogeneity of neurofibromatosis type 1 in a large international registry". JCI Insight. 5 (16). doi:10.1172/jci.insight.136262. PMC 7455126. PMID 32814709. S2CID 221202508.
- Hirbe AC, Gutmann DH (August 2014). "Neurofibromatosis type 1: a multidisciplinary approach to care". The Lancet. Neurology. 13 (8): 834–843. doi:10.1016/s1474-4422(14)70063-8. PMID 25030515. S2CID 9813111.
- Raue F (2012-12-11). "Faculty Opinions recommendation of Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1". Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature. doi:10.3410/f.717964914.793467319. Retrieved 2022-07-14.
- SergottRC (2007-03-23). "Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations". Yearbook of Ophthalmology. 2007: 200–201. doi:10.1016/s0084-392x(08)70156-4. ISSN 0084-392X.
- Torres Nupan MM, Velez Van Meerbeke A, López Cabra CA, Herrera Gomez PM (2017). "Cognitive and Behavioral Disorders in Children with Neurofibromatosis Type 1". Frontiers in Pediatrics. 5: 227. doi:10.3389/fped.2017.00227. PMC 5670111. PMID 29164079.
- Ostendorf AP, Gutmann DH, Weisenberg JL (October 2013). "Epilepsy in individuals with neurofibromatosis type 1". Epilepsia. 54 (10): 1810–1814. doi:10.1111/epi.12348. PMID 24032542. S2CID 1603461.
- Terry AR, Jordan JT, Schwamm L, Plotkin SR (January 2016). "Increased Risk of Cerebrovascular Disease Among Patients With Neurofibromatosis Type 1: Population-Based Approach". Stroke. 47 (1): 60–65. doi:10.1161/strokeaha.115.011406. PMID 26645253. S2CID 3253811.
- Bachir S, Shah S, Shapiro S, Koehler A, Mahammedi A, Samy RN; et al. (2021). "Neurofibromatosis Type 2 (NF2) and the Implications for Vestibular Schwannoma and Meningioma Pathogenesis". Int J Mol Sci. 22 (2). doi:10.3390/ijms22020690. PMC 7828193. PMID 33445724.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, Lalloo F (February 2010). "Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service". American Journal of Medical Genetics. Part A. 152A (2): 327–332. doi:10.1002/ajmg.a.33139. PMID 20082463. S2CID 25884728.
- Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, et al. (December 1992). "A genetic study of type 2 neurofibromatosis in the United Kingdom. II. Guidelines for genetic counselling". Journal of Medical Genetics. 29 (12): 847–852. doi:10.1136/jmg.29.12.847. PMC 1016199. PMID 1479599. S2CID 10548080.
- Evans DG (June 2009). "Neurofibromatosis type 2 (NF2): a clinical and molecular review". Orphanet Journal of Rare Diseases. 4 (1): 16. doi:10.1186/1750-1172-4-16. PMC 2708144. PMID 19545378. S2CID 15410658.
- Evans DG, Hartley CL, Smith PT, King AT, Bowers NL, Tobi S, et al. (January 2020). "Incidence of mosaicism in 1055 de novo NF2 cases: much higher than previous estimates with high utility of next-generation sequencing". Genetics in Medicine. 22 (1): 53–59. doi:10.1038/s41436-019-0598-7. PMID 31273341. S2CID 195795815.
- Halliday D, Emmanouil B, Vassallo G, Lascelles K, Nicholson J, Chandratre S, et al. (August 2019). "Trends in phenotype in the English paediatric neurofibromatosis type 2 cohort stratified by genetic severity". Clinical Genetics. 96 (2): 151–162. doi:10.1111/cge.13551. hdl:10026.1/15073. PMID 30993672. S2CID 119105041.
- Asthagiri AR, Parry DM, Butman JA, Kim HJ, Tsilou ET, Zhuang Z, Lonser RR (June 2009). "Neurofibromatosis type 2". Lancet. 373 (9679): 1974–1986. doi:10.1016/S0140-6736(09)60259-2. PMC 4748851. PMID 19476995.
- Dirks MS, Butman JA, Kim HJ, Wu T, Morgan K, Tran AP, et al. (July 2012). "Long-term natural history of neurofibromatosis Type 2-associated intracranial tumors". Journal of Neurosurgery. 117 (1): 109–117. doi:10.3171/2012.3.jns111649. PMC 4749021. PMID 22503123.
- Baser ME, Friedman JM, Aeschliman D, Joe H, Wallace AJ, Ramsden RT, Evans DG (October 2002). "Predictors of the risk of mortality in neurofibromatosis 2". American Journal of Human Genetics. 71 (4): 715–723. doi:10.1086/342716. PMC 378530. PMID 12235555. S2CID 23250053.
- Henske, Elizabeth P.; Jóźwiak, Sergiusz; Kingswood, J. Christopher; Sampson, Julian R.; Thiele, Elizabeth A. (2016-05-26). "Tuberous sclerosis complex". Nature Reviews. Disease Primers. 2: 16035. doi:10.1038/nrdp.2016.35. ISSN 2056-676X. PMID 27226234. S2CID 68051262.
- Zoncu, Roberto; Efeyan, Alejo; Sabatini, David M. (2010-12-15). "mTOR: from growth signal integration to cancer, diabetes and ageing". Nature Reviews. Molecular Cell Biology. 12 (1): 21–35. doi:10.1038/nrm3025. ISSN 1471-0080. PMC 3390257. PMID 21157483.
- Curatolo, Paolo; Bombardieri, Roberta; Jozwiak, Sergiusz (2008-08-23). "Tuberous sclerosis". The Lancet. 372 (9639): 657–668. doi:10.1016/S0140-6736(08)61279-9. ISSN 0140-6736. PMID 18722871. S2CID 28589689.
- Stafstrom, Carl E.; Staedtke, Verena; Comi, Anne M. (2017). "Epilepsy Mechanisms in Neurocutaneous Disorders: Tuberous Sclerosis Complex, Neurofibromatosis Type 1, and Sturge-Weber Syndrome". Frontiers in Neurology. 8: 87. doi:10.3389/fneur.2017.00087. ISSN 1664-2295. PMC 5355446. PMID 28367137.
- Chu-Shore, Catherine J.; Major, Philippe; Camposano, Susana; Muzykewicz, David; Thiele, Elizabeth A. (2010-07-01). "The natural history of epilepsy in tuberous sclerosis complex". Epilepsia. 51 (7): 1236–1241. doi:10.1111/j.1528-1167.2009.02474.x. ISSN 1528-1167. PMC 3065368. PMID 20041940.
- Northrup, Hope; Krueger, Darcy A.; International Tuberous Sclerosis Complex Consensus Group (2013-10-01). "Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference". Pediatric Neurology. 49 (4): 243–254. doi:10.1016/j.pediatrneurol.2013.08.001. ISSN 1873-5150. PMC 4080684. PMID 24053982.
- Roth, Jonathan; Roach, E. Steve; Bartels, Ute; Jóźwiak, Sergiusz; Koenig, Mary Kay; Weiner, Howard L.; Franz, David N.; Wang, Henry Z. (2013-10-17). "Subependymal giant cell astrocytoma: diagnosis, screening, and treatment. Recommendations from the International Tuberous Sclerosis Complex Consensus Conference 2012". Pediatric Neurology. 49 (6): 439–444. doi:10.1016/j.pediatrneurol.2013.08.017. ISSN 1873-5150. PMID 24138953.
- de Vries, Petrus J.; Whittemore, Vicky H.; Leclezio, Loren; Byars, Anna W.; Dunn, David; Ess, Kevin C.; Hook, Dena; King, Bryan H.; Sahin, Mustafa; Jansen, Anna (2015-01-01). "Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist". Pediatric Neurology. 52 (1): 25–35. doi:10.1016/j.pediatrneurol.2014.10.004. ISSN 1873-5150. PMC 4427347. PMID 25532776.
- Adriaensen, M. E. a. P. M.; Schaefer-Prokop, C. M.; Duyndam, D. a. C.; Zonnenberg, B. A.; Prokop, M. (2011-07-01). "Radiological evidence of lymphangioleiomyomatosis in female and male patients with tuberous sclerosis complex". Clinical Radiology. 66 (7): 625–628. doi:10.1016/j.crad.2011.02.009. ISSN 1365-229X. PMID 21459371.
- Ewalt, D. H.; Sheffield, E.; Sparagana, S. P.; Delgado, M. R.; Roach, E. S. (1998-07-01). "Renal lesion growth in children with tuberous sclerosis complex". The Journal of Urology. 160 (1): 141–145. doi:10.1016/S0022-5347(01)63072-6. ISSN 0022-5347. PMID 9628635.
- Borkowska, Julita; Schwartz, Robert A.; Kotulska, Katarzyna; Jozwiak, Sergiusz (2011-01-01). "Tuberous sclerosis complex: tumors and tumorigenesis". International Journal of Dermatology. 50 (1): 13–20. doi:10.1111/j.1365-4632.2010.04727.x. ISSN 1365-4632. PMID 21182496. S2CID 34212098.
- Jóźwiak, Sergiusz; Kotulska, Katarzyna; Kasprzyk-Obara, Jolanta; Domańska-Pakieła, Dorota; Tomyn-Drabik, Małgorzata; Roberts, Penelope; Kwiatkowski, David (2006-10-01). "Clinical and genotype studies of cardiac tumors in 154 patients with tuberous sclerosis complex". Pediatrics. 118 (4): e1146–1151. doi:10.1542/peds.2006-0504. ISSN 1098-4275. PMID 16940165. S2CID 25133016.
- Teng, Joyce M. C.; Cowen, Edward W.; Wataya-Kaneda, Mari; Gosnell, Elizabeth S.; Witman, Patricia M.; Hebert, Adelaide A.; Mlynarczyk, Greg; Soltani, Keyoumars; Darling, Thomas N. (2014-10-01). "Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements". JAMA Dermatology. 150 (10): 1095–1101. doi:10.1001/jamadermatol.2014.938. ISSN 2168-6084. PMID 25029267.
- Sudarsanam, Annapurna; Ardern-Holmes, Simone L. (2013-11-25). "Sturge–Weber syndrome: From the past to the present". European Journal of Paediatric Neurology. 18 (3): 257–266. doi:10.1016/j.ejpn.2013.10.003. ISSN 1090-3798. PMID 24275166.
- Shirley, Matthew D.; Tang, Hao; Gallione, Carol J.; Baugher, Joseph D.; Frelin, Laurence P.; Cohen, Bernard; North, Paula E.; Marchuk, Douglas A.; Comi, Anne M.; Pevsner, Jonathan (2013-05-23). "Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ". The New England Journal of Medicine. 368 (21): 1971–1979. doi:10.1056/NEJMoa1213507. ISSN 1533-4406. PMC 3749068. PMID 23656586.
- Ch'ng, Sydney; Tan, Swee T. (2007-07-09). "Facial port-wine stains - clinical stratification and risks of neuro-ocular involvement". Journal of Plastic, Reconstructive & Aesthetic Surgery: JPRAS. 61 (8): 889–893. doi:10.1016/j.bjps.2007.05.011. ISSN 1878-0539. PMID 17604243.
- Waelchli, R.; Aylett, S.E.; Robinson, K.; Chong, W.K.; Martinez, A.E.; Kinsler, V.A. (2014-06-27). "New vascular classification of port‐wine stains: improving prediction of Sturge–Weber risk". British Journal of Dermatology. 171 (4): 861–867. doi:10.1111/bjd.13203. ISSN 0007-0963. PMC 4284033. PMID 24976116.
- Sabeti, Sara; Ball, Karen L.; Burkhart, Craig; Eichenfield, Lawrence; Fernandez Faith, Esteban; Frieden, Ilona J.; Geronemus, Roy; Gupta, Deepti; Krakowski, Andrew C.; Levy, Moise L.; Metry, Denise (2021-01-01). "Consensus Statement for the Management and Treatment of Port-Wine Birthmarks in Sturge-Weber Syndrome". JAMA Dermatology. 157 (1): 98–104. doi:10.1001/jamadermatol.2020.4226. ISSN 2168-6068. PMC 8547264. PMID 33175124.
- Lambiase, Alessandro; Mantelli, Flavio; Bruscolini, Alice; La Cava, Maurizio; Abdolrahimzadeh, Solmaz (2016-05-13). "Ocular manifestations of Sturge-Weber syndrome: pathogenesis, diagnosis, and management". Clinical Ophthalmology. 10: 871–878. doi:10.2147/opth.s101963. ISSN 1177-5483. PMC 4874637. PMID 27257371.
- Silverstein, Marlee; Salvin, Jonathan (2019-09-01). "Ocular manifestations of Sturge–Weber syndrome". Current Opinion in Ophthalmology. 30 (5): 301–305. doi:10.1097/icu.0000000000000597. ISSN 1040-8738. PMID 31313748. S2CID 197423353.
- Higueros, E.; Roe, E.; Granell, E.; Baselga, E. (2017-06-01). "Sturge-Weber Syndrome: A Review". Actas Dermo-Sifiliograficas. 108 (5): 407–417. doi:10.1016/j.ad.2016.09.022. ISSN 1578-2190. PMID 28126187.
- Singh, Arun D.; Kaiser, Peter K.; Sears, Jonathan E. (2005-03-01). "Choroidal hemangioma". Ophthalmology Clinics of North America. 18 (1): 151–161, ix. doi:10.1016/j.ohc.2004.07.004. ISSN 0896-1549. PMID 15763200.
- Powell, Sebastian; Fosi, Tangunu; Sloneem, Jenny; Hawkins, Christina; Richardson, Hanna; Aylett, Sarah (2021-09-01). "Neurological presentations and cognitive outcome in Sturge-Weber syndrome". European Journal of Paediatric Neurology. 34: 21–32. doi:10.1016/j.ejpn.2021.07.005. ISSN 1090-3798. PMID 34293629.
- Sujansky, Eva; Conradi, Susan (1995-05-22). "Outcome of Sturge-Weber syndrome in 52 adults". American Journal of Medical Genetics. 57 (1): 35–45. doi:10.1002/ajmg.1320570110. ISSN 0148-7299. PMID 7645596.
- Sugano, Hidenori; Iimura, Yasushi; Igarashi, Ayuko; Nakazawa, Mika; Suzuki, Hiroharu; Mitsuhashi, Takumi; Nakajima, Madoka; Higo, Takuma; Ueda, Tetsuya; Nakanishi, Hajime; Niijima, Shinichi (2021-04-01). "Extent of Leptomeningeal Capillary Malformation is Associated With Severity of Epilepsy in Sturge-Weber Syndrome". Pediatric Neurology. 117: 64–71. doi:10.1016/j.pediatrneurol.2020.12.012. ISSN 0887-8994. PMID 33677229. S2CID 232140769.
- Frank, Nicole Alexandra; Greuter, Ladina; Dill, Patricia Elsa; Guzman, Raphael; Soleman, Jehuda (2022-05-01). "Focal lesionectomy as surgical treatment of epilepsy in patients with Sturge-Weber syndrome: a case-based systematic review and meta-analysis". Neurosurgical Focus. 52 (5): E4. doi:10.3171/2022.2.focus21788. ISSN 1092-0684. PMID 35535828. S2CID 248595880.
- Pascual-Castroviejo, Ignacio; Pascual-Pascual, Samuel-Ignacio; Velazquez-Fragua, Ramón; Viaño, Juán (2008-07-01). "Sturge-Weber syndrome: study of 55 patients". The Canadian Journal of Neurological Sciences. Le Journal Canadien des Sciences Neurologiques. 35 (3): 301–307. doi:10.1017/s0317167100008878. ISSN 0317-1671. PMID 18714797. S2CID 35525783.
- Kaelin, William G. (2002-09-01). "Molecular basis of the VHL hereditary cancer syndrome". Nature Reviews Cancer. 2 (9): 673–682. doi:10.1038/nrc885. ISSN 1474-175X. PMID 12209156. S2CID 20186415.
- Sgambati, M.T.; Stolle, C.; Choyke, P.L.; Walther, M.M.; Zbar, B.; Linehan, W.M.; Glenn, G.M. (2000-01-01). "Mosaicism in von Hippel–Lindau Disease: Lessons from Kindreds with Germline Mutations Identified in Offspring with Mosaic Parents". The American Journal of Human Genetics. 66 (1): 84–91. doi:10.1086/302726. ISSN 0002-9297. PMC 1288351. PMID 10631138.
- Wilding, Anna; Ingham, Sarah Louise; Lalloo, Fiona; Clancy, Tara; Huson, Susan M; Moran, Anthony; Evans, D Gareth (2012-02-23). "Life expectancy in hereditary cancer predisposing diseases: an observational study". Journal of Medical Genetics. 49 (4): 264–269. doi:10.1136/jmedgenet-2011-100562. ISSN 0022-2593. PMID 22362873. S2CID 41306596.
- Maher, Eamonn R.; Neumann, Hartmut Ph; Richard, Stéphane (2011-03-09). "von Hippel-Lindau disease: a clinical and scientific review". European Journal of Human Genetics: EJHG. 19 (6): 617–623. doi:10.1038/ejhg.2010.175. ISSN 1476-5438. PMC 3110036. PMID 21386872.
- Lonser, Russell R.; Butman, John A.; Huntoon, Kristin; Asthagiri, Ashok R.; Wu, Tianxia; Bakhtian, Kamran D.; Chew, Emily Y.; Zhuang, Zhengping; Linehan, W. Marston; Oldfield, Edward H. (2014-05-01). "Prospective natural history study of central nervous system hemangioblastomas in von Hippel-Lindau disease". Journal of Neurosurgery. 120 (5): 1055–1062. doi:10.3171/2014.1.JNS131431. ISSN 1933-0693. PMC 4762041. PMID 24579662.
- Dollfus, Hélène; Massin, Pascale; Taupin, Pierre; Nemeth, Catherine; Amara, Sandrine; Giraud, Sophie; Béroud, Christophe; Dureau, Pascal; Gaudric, Alain; Landais, Paul; Richard, Stéphane (2002-09-01). "Retinal hemangioblastoma in von Hippel-Lindau disease: a clinical and molecular study". Investigative Ophthalmology & Visual Science. 43 (9): 3067–3074. ISSN 0146-0404. PMID 12202531.
- Kreusel, Klaus-Martin; Bechrakis, Nikolaos E.; Krause, Lothar; Neumann, Hartmut P. H.; Foerster, Michael H. (2006-08-01). "Retinal angiomatosis in von Hippel-Lindau disease: a longitudinal ophthalmologic study". Ophthalmology. 113 (8): 1418–1424. doi:10.1016/j.ophtha.2006.02.059. ISSN 1549-4713. PMID 16769118.
- Wittström, Elisabeth; Nordling, Margareta; Andréasson, Sten (2014-02-20). "Genotype-phenotype correlations, and retinal function and structure in von Hippel-Lindau disease". Ophthalmic Genetics. 35 (2): 91–106. doi:10.3109/13816810.2014.886265. ISSN 1744-5094. PMID 24555745. S2CID 20618502.
- Toy, Brian C.; Agrón, Elvira; Nigam, Divya; Chew, Emily Y.; Wong, Wai T. (2012-12-01). "Longitudinal analysis of retinal hemangioblastomatosis and visual function in ocular von Hippel-Lindau disease". Ophthalmology. 119 (12): 2622–2630. doi:10.1016/j.ophtha.2012.06.026. ISSN 1549-4713. PMC 3504630. PMID 22906772.
- Walther, M. M.; Reiter, R.; Keiser, H. R.; Choyke, P. L.; Venzon, D.; Hurley, K.; Gnarra, J. R.; Reynolds, J. C.; Glenn, G. M.; Zbar, B.; Linehan, W. M. (1999-09-01). "Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma". The Journal of Urology. 162 (3 Pt 1): 659–664. doi:10.1097/00005392-199909010-00004. ISSN 0022-5347. PMID 10458336.
- Duffey, Branden G.; Choyke, Peter L.; Glenn, Gladys; Grubb, Robert L.; Venzon, David; Linehan, W. Marston; Walther, McClellan M. (2004-07-01). "The relationship between renal tumor size and metastases in patients with von Hippel-Lindau disease". The Journal of Urology. 172 (1): 63–65. doi:10.1097/01.ju.0000132127.79974.3f. ISSN 0022-5347. PMID 15201738.
- Steinbach, F.; Novick, A. C.; Zincke, H.; Miller, D. P.; Williams, R. D.; Lund, G.; Skinner, D. G.; Esrig, D.; Richie, J. P.; deKernion, J. B. (1995-06-01). "Treatment of renal cell carcinoma in von Hippel-Lindau disease: a multicenter study". The Journal of Urology. 153 (6): 1812–1816. doi:10.1016/S0022-5347(01)67318-X. ISSN 0022-5347. PMID 7752324.
- Binderup, Marie Louise Mølgaard; Bisgaard, Marie Luise; Harbud, Vibeke; Møller, Hans Ulrik; Gimsing, Steen; Friis-Hansen, Lennart; Hansen, Thomas van Overeem; Bagi, Per; Knigge, Ulrich; Kosteljanetz, Michael; Bøgeskov, Lars (2013-12-01). "Von Hippel-Lindau disease (vHL). National clinical guideline for diagnosis and surveillance in Denmark. 3rd edition". Danish Medical Journal. 60 (12): B4763. ISSN 2245-1919. PMID 24355456.
- Charlesworth, Michael; Verbeke, Caroline S.; Falk, Gavin A.; Walsh, Matthew; Smith, Andrew M.; Morris-Stiff, Gareth (2012-02-28). "Pancreatic lesions in von Hippel-Lindau disease? A systematic review and meta-synthesis of the literature". Journal of Gastrointestinal Surgery. 16 (7): 1422–1428. doi:10.1007/s11605-012-1847-0. ISSN 1873-4626. PMID 22370733. S2CID 2090370.
- Blansfield, Joseph A.; Choyke, Lynda; Morita, Shane Y.; Choyke, Peter L.; Pingpank, James F.; Alexander, H. Richard; Seidel, Geoffrey; Shutack, Yvonne; Yuldasheva, Nargiza; Eugeni, Michelle; Bartlett, David L. (2007-12-01). "Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs)". Surgery. 142 (6): 814–818, discussion 818.e1–2. doi:10.1016/j.surg.2007.09.012. ISSN 1532-7361. PMC 6771023. PMID 18063061.
- Radtke, Heather B.; Lalor, Leah E.; Basel, Donald G.; Siegel, Dawn H. (2020-11-18). "Clinical Implications of Mosaicism and Low-Level Mosaicism in Neurocutaneous Disorders". Current Genetic Medicine Reports. 8 (4): 132–139. doi:10.1007/s40142-020-00193-9. ISSN 2167-4876. S2CID 226985730.
- Lalonde, Emilie; Ebrahimzadeh, Jessica; Rafferty, Keith; Richards‐Yutz, Jennifer; Grant, Richard; Toorens, Erik; Marie Rosado, Jennifer; Schindewolf, Erica; Ganguly, Tapan; Kalish, Jennifer M.; Deardorff, Matthew A. (2019-02-13). "Molecular diagnosis of somatic overgrowth conditions: A single‐center experience". Molecular Genetics & Genomic Medicine. 7 (3): e536. doi:10.1002/mgg3.536. ISSN 2324-9269. PMC 6418364. PMID 30761771.
- McNulty, Samantha N.; Evenson, Michael J.; Corliss, Meagan M.; Love-Gregory, Latisha D.; Schroeder, Molly C.; Cao, Yang; Lee, Yi-Shan; Drolet, Beth A.; Neidich, Julie A.; Cottrell, Catherine E.; Heusel, Jonathan W. (2019-10-03). "Diagnostic Utility of Next-Generation Sequencing for Disorders of Somatic Mosaicism: A Five-Year Cumulative Cohort". The American Journal of Human Genetics. 105 (4): 734–746. doi:10.1016/j.ajhg.2019.09.002. ISSN 0002-9297. PMC 6817554. PMID 31585106.
- Spits, C.; De Rycke, M.; Van Ranst, N.; Joris, H.; Verpoest, W.; Lissens, W.; Devroey, P.; Van Steirteghem, A.; Liebaers, I.; Sermon, K. (2005-05-01). "Preimplantation genetic diagnosis for neurofibromatosis type 1". Molecular Human Reproduction. 11 (5): 381–387. doi:10.1093/molehr/gah170. ISSN 1360-9947. PMID 15833774.
- Bodemer, C.; Diociaiuti, A.; Hadj‐Rabia, S.; Robert, M.P.; Desguerre, I.; Manière, M.‐C.; Dure‐Molla, M.; De Liso, P.; Federici, M.; Galeotti, A.; Fusco, F. (2020-07-17). "Multidisciplinary consensus recommendations from a European network for the diagnosis and practical management of patients with incontinentia pigmenti". Journal of the European Academy of Dermatology and Venereology. 34 (7): 1415–1424. doi:10.1111/jdv.16403. ISSN 0926-9959. PMID 32678511. S2CID 220610122.
- Fox, Jonah; Ben-Shachar, Shay; Uliel, Shimrit; Svirsky, Ran; Saitsu, Hirotomo; Matsumoto, Naomichi; Fattal-Valevski, Aviva (2017-01-27). "Rare familial TSC2 gene mutation associated with atypical phenotype presentation of Tuberous Sclerosis Complex". American Journal of Medical Genetics Part A. 173 (3): 744–748. doi:10.1002/ajmg.a.38027. ISSN 1552-4825. PMID 28127866. S2CID 6584102.
- Northrup, Hope; Krueger, Darcy A.; International Tuberous Sclerosis Complex Consensus Group (2013-10-01). "Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference". Pediatric Neurology. 49 (4): 243–254. doi:10.1016/j.pediatrneurol.2013.08.001. ISSN 1873-5150. PMC 4080684. PMID 24053982.
- Stewart, Douglas R.; Korf, Bruce R.; Nathanson, Katherine L.; Stevenson, David A.; Yohay, Kaleb (2018-07-01). "Care of adults with neurofibromatosis type 1: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG)". Genetics in Medicine. 20 (7): 671–682. doi:10.1038/gim.2018.28. ISSN 1098-3600. PMID 30006586. S2CID 49721557.
- Amin, S.; Kingswood, J. C.; Bolton, P. F.; Elmslie, F.; Gale, D. P.; Harland, C.; Johnson, S. R.; Parker, A.; Sampson, J. R.; Smeaton, M.; Wright, I. (2019-03-01). "The UK guidelines for management and surveillance of Tuberous Sclerosis Complex". QJM: Monthly Journal of the Association of Physicians. 112 (3): 171–182. doi:10.1093/qjmed/hcy215. ISSN 1460-2393. PMID 30247655.
- Wilson, Britney N.; John, Ann M.; Handler, Marc Zachary; Schwartz, Robert A. (2021-06-01). "Neurofibromatosis type 1: New developments in genetics and treatment". Journal of the American Academy of Dermatology. 84 (6): 1667–1676. doi:10.1016/j.jaad.2020.07.105. ISSN 0190-9622. PMID 32771543. S2CID 221092583.
- Grossen, Audrey; Gavula, Theresa; Chrusciel, Deepti; Evans, Alexander; McNall-Knapp, Rene; Taylor, Ashley; Fossey, Benay; Brakefield, Margaret; Carter, Carrick; Schwartz, Nadine; Gross, Naina (2022-05-01). "Multidisciplinary neurocutaneous syndrome clinics: a systematic review and institutional experience". Neurosurgical Focus. 52 (5): E2. doi:10.3171/2022.2.focus21776. ISSN 1092-0684. PMID 35535824. S2CID 248499994.
External links
- OMIM is an Online Catalog of Human Genes and Genetic Disorders