XXXY syndrome
XXXY syndrome is a genetic condition characterized by a sex chromosome aneuploidy, where individuals have two extra X chromosomes.[2] People in most cases have two sex chromosomes: an X and a Y or two X chromosomes. The presence of one Y chromosome with a functioning SRY gene causes the expression of genes that determine maleness. Because of this, XXXY syndrome only affects males. The additional two X chromosomes in males with XXXY syndrome causes them to have 48 chromosomes, instead of the typical 46. XXXY syndrome is therefore often referred to as 48,XXXY. There is a wide variety of symptoms associated with this syndrome, including cognitive and behavioral problems, taurodontism, and infertility.[2][3] This syndrome is usually inherited via a new mutation in one of the parents' gametes, as those affected by it are usually infertile. It is estimated that XXXY affects one in every 50,000 male births.[3]
| XXXY syndrome | |
|---|---|
| Other names | 48,XXXY syndrome[1] |
Signs and symptoms
The symptoms of 48,XXXY syndrome are similar to those of Klinefelter syndrome, though the symptoms are usually more severe in 48,XXXY syndrome. Like Klinefelter syndrome, the presence of additional X chromosomes affects the male reproductive system, can cause physical abnormalities, and can affect cognitive development. When comparing 47,XXY and 48,XXXY, there is a greater risk for congenital malformations and more medical problems in those with 48,XXXY.[3]
Reproductive
Those with XXXY syndrome can have testicular dysgenesis and hypergonadotrophic hypogonadism.[3] Testicular dysgenesis is a condition in which a male has incomplete or complete loss of spermatogenesis, so that the individual produces very low levels, or no sperm.[4] This results in infertility of that individual.[4] Hypergonadotrophic hypogonadism is a condition in which the function of the testes in males is reduced and can result in low levels of sex steroids produced like testosterone.
Physical
Males with 48,XXXY can have average or tall stature, which becomes more prominent in adulthood. Facial dysmorphism is common in males with 48,XXXY and can include increased distance between the eyes (hypertelorism), skin folds of the upper eyelid (epicanthal folds), up-slanting opening between the eyelids (palpebral fissures) and hooded eyelids. Other physical features include the fifth finger or "pinkie" to be bent inwards towards the fourth finger (clinodactyly), short nail beds, flat feet, double jointedness (hyperextensibility) and prominent elbows with cubitus varus where the arm rests closer to the body. Musculoskeletal features may include congenital elbow dislocation and the limited ability of the feet to roll inwards while walking and upon landing.[3] Micropenis is another common symptom of this syndrome.[5]
Individuals affected with XXXY are also prone to developing taurodontism, which often presents early in life, and can be an early indicator of XXY syndrome.[2] Those with this syndrome are also prone to hip dysplasia, and other joint abnormalities.[6] An individual’s symptoms vary due to differing androgen deficiencies, and also with alter with age. Prepubescent boys with XXXY syndrome may not differ in physical appearance from a child without the syndrome. This is likely because androgen levels do not differ among pre-pubescent boys, but a difference does arise as puberty progresses.[2] Those with XXXY syndrome may also experience feminine distribution of adipose tissue, and gynecomastia may also be present.[2] Tall stature is more likely to appear in adolescence, when androgen levels begin to differ between those with XXXY syndrome and those that do not have it.
Cognitive and developmental
Neurological effects are believed to be more severe as the number of extra X chromosomes increases; a male with 48,XXXY is likely to have more severe symptoms than a male with Klinefelter syndrome.[5] Developmental delays are common in infancy and childhood. Expected symptoms include speech delays, motor delays, and hypotonia (lack of muscle tone), also known as floppy baby syndrome. Individuals with XXXY syndrome exhibit cognitive and behavioral problems.[2]
Patients typically show altered adaptive behavior, which is the ability of an individual to demonstrate essential living skills, including: social skills, community living, safety, functional use of academic skills and self-care. People with XXXY syndrome were found to score significantly less in the domains of daily living skills and communication compared to XXYY, and XXY individuals.[3] This means that they typically demonstrate little ability in the domains of self-care, social skills, safety, application of academic skills, and responsibility.[3]
Individuals with this syndrome also experience emotional symptoms such as anxiety symptoms, obsessive-compulsive behaviors, behavioral dysregulational and emotional immaturity.[3] People with this syndrome typically have an IQ in the range of 40-60, where the average IQ range is 95-110.[5][7] They also experience language-based learning disabilities that can affect their communication with others.[4] Those with XXXY syndrome tend to display less externalizing and internalizing behaviors compared to those with 48,XXYY syndrome, which may have a positive effect on their social functioning.[8] These individuals may also have increased vulnerability for autistic features.[8] Changes in testosterone as well as androgen deficits may contribute to these individuals’ social behaviors that put them at increased risk for autistic features.[8]
Cause
The cause of 48,XXXY can be from non-disjunction in the paternal sperm or non-disjunction in the maternal oocyte.[3] The most likely scenario for the existence of this aneuploidy is that each party (maternal and paternal) equally contributed to it, by the egg giving an XX and the sperm giving an XY.[3]
In the case where the sperm is the genetic cause of 48,XXXY syndrome, the sperm would have to contain two X chromosomes and one Y chromosome. This would be caused by two nondisjunction events in spermatogenesis, both meiosis I and meiosis II. The duplicated X chromosome in the sperm would have to fail to separate in both meiosis I and meiosis II for a sperm as well as the X and Y chromosomes would have to be in the same sperm. Then the XXY sperm would fertilize a normal oocyte to make a XXXY zygote.[5]
In the case where the oocyte is the genetic cause of 48,XXXY syndrome the oocyte would contain three X chromosome. This would be caused by two nondisjunction events during oogenesis. In meiosis I both sets of duplicated X chromosomes would have to be not separated. Then in meiosis II one set of X chromosomes would have to not separate and the other set would separate resulting in one oocyte with three X chromosomes. A normal sperm containing a Y chromosome would have to fertilize the XXX oocyte to make a XXXY zygote.[5]
Diagnosis
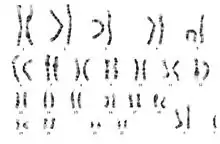
Diagnosis of 48,XXXY is usually done by a standard karyotype.[3] A karyotype is a chromosomal analysis in which a full set of chromosomes can be seen for an individual. The presence of the additional 2 X chromosomes on the karyotype are indicative of XXXY syndrome.
Another way to diagnosis 48,XXXY is by chromosomal microarray showing the presence of extra X chromosomes.[3] Chromosomal microarray (CMA) is used to detect extra or missing chromosomal segments or whole chromosomes. CMA uses microchip-based testing to analyze many pieces of DNA. Males with 48,XXXY are diagnosed anywhere from before birth to adulthood as a result of the range in the severity of symptoms.[3] The age range at diagnosis is likely due to the fact that XXXY is a rare syndrome, and does not cause as extreme phenotypes as other variants of Klinefelter syndrome (such as XXXXY).[3]
Diagnostic testing could also be done via blood samples. Elevated levels of follicle stimulating hormone, luteinizing hormone, and low levels of testosterone can be indicative of this syndrome.[5]
Management
Treatment
Treatments exist for the various symptoms associated with XXXY syndrome. Testosterone therapy, which is giving affected individuals doses of testosterone on a regular basis, has been shown to reduce aggressive behavior in these patients.[3] However, this therapy has also been associated with negative side effects: worsening of behavior, and osteoporosis.[3] Not all individuals are applicable for testosterone therapy, as the best results are often achieved when dosage begins at the initiation of puberty, and these individuals are often diagnosed at a later age, or not at all.[3] Testosterone therapy has been shown to have no positive effect on fertility.[2]
Consideration of the psychological phenotype of individuals with XXXY should be taken into account when treating these patients, because these traits affect compliance with treatments.[3] When caught early, Taurodontism can be treated with a root canal and is often successful.[2] Appropriate planning to avoid Taurodontism is possible, but this syndrome must be diagnosed early, which is not common.[2] Taurodontism can often be detected as a symptom of XXXY syndrome before other characteristics develop, and can be an early indicator for it. Surgical treatments to correct joint problems, such as hip dysplasia are common, and are often successful alongside physiotherapy.[6]
Those with XXXY syndrome can also attend speech therapy.[5] This form of therapy helps patients to understand and produce more complex language.[5] Those with XXXY syndrome tend to experience more severe speech delays, so this form of treatment can be very beneficial to them, and can help them to communicate better with other people.[5]
Since hypotonia is common in those with this syndrome, physical therapy can also be helpful.[5] This form of therapy may help these individuals develop muscle tone, and increase balance and coordination.[5]
Quality of life
In mild cases, individuals with XXXY syndrome may lead a relatively good life. These individuals may face difficulties in communicating with others due to their language-based deficits. These deficits may make forming bonds with others difficult, but fulfilling relationships with others are still achievable. Those with higher scores in adaptive functioning are likely to have higher quality of life because they can be independent.[3]
Notable people
- Caroline Cossey, British model[9]
References
- "XXXY syndrome". The Genetic and Rare Diseases Information Center (GARD). NIH. 10 August 2016. Retrieved 19 March 2019.
- Joseph, Michael (2008-05-01). "Endodontic treatment in three taurodontic teeth associated with 48,XXXY Klinefelter syndrome: a review and case report". Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontology. 105 (5): 670–677. doi:10.1016/j.tripleo.2007.11.015. PMID 18442747.
- Tartaglia, Nicole; Ayari, Natalie; Howell, Susan; D’Epagnier, Cheryl; Zeitler, Philip (2011-06-01). "48,XXYY, 48,XXXY and 49,XXXXY syndromes: not just variants of Klinefelter syndrome". Acta Paediatrica. 100 (6): 851–860. doi:10.1111/j.1651-2227.2011.02235.x. ISSN 1651-2227. PMC 3314712. PMID 21342258.
- Skakkebæk, N.E.; Meyts, E. Rajpert-De; Main, K.M. (2001-05-01). "Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects: Opinion". Human Reproduction. 16 (5): 972–978. doi:10.1093/humrep/16.5.972. ISSN 0268-1161. PMID 11331648.
- Visootsak, Jeannie; Graham, John M. (2006-10-24). "Klinefelter syndrome and other sex chromosomal aneuploidies". Orphanet Journal of Rare Diseases. 1: 42. doi:10.1186/1750-1172-1-42. ISSN 1750-1172. PMC 1634840. PMID 17062147.
- Simşek, P. O.; Utine, G. E.; Alikaşifoğlu, A.; Alanay, Y.; Boduroğlu, K.; Kandemir, N. (2009). "Rare sex chromosome aneuploidies: 49,XXXXY and 48,XXXY syndromes". The Turkish Journal of Pediatrics. 51 (3): 294–297. ISSN 0041-4301. PMID 19817277.
- "APA PsycNet".
- Edgren, Gretchen (September 1991). "The transformation of Tula (transsexual Caroline Cossey)". Playboy. 38 (9): 102. Archived from the original on 6 December 2004.
Further reading
- Ferrier, Pierre E. (1974-01-01). "The XXXY Klinefelter Syndrome in Childhood". Archives of Pediatrics & Adolescent Medicine. 127 (1): 104–5. doi:10.1001/archpedi.1974.02110200106016. ISSN 0002-922X. PMID 4809784.