Ring chromosome 20 syndrome
Ring chromosome 20, ring-shaped chromosome 20 or r(20) syndrome is a rare human chromosome abnormality where the two arms of chromosome 20 fuse to form a ring chromosome. The syndrome is associated with epileptic seizures, behaviour disorders and intellectual disability.[1]
| Ring chromosome 20 syndrome | |
|---|---|
_-_ring_chromosome_20.PNG.webp) | |
| Normal chromosome 20 and ring chromosome 20 in an heterozygote patient |
When not all cells contain a ring chromosome 20, the individual has ring 20 chromosomal mosaicism. Ring Chromosome 20 syndrome is thought to be an underdiagnosed condition. Since chromosomal analysis or karyotype testing is not a routine investigation for patients with epilepsy, the diagnosis of ring chromosome 20 syndrome is typically delayed or unrecognized.
Presentation
Recurrent seizures are the most recognizable feature of this syndrome and are most often the first sign of this syndrome. These syndromes are often ongoing and poorly responsive to anti-seizure medications.[2][3] Most patients develop seizures the first few years of life, but the age of onset ranges from ages 1 to 17. Different types of seizure have been reported in this syndrome. The most common seizure type appears to be brief focal onset epileptic seizures with impairment of consciousness and awareness, known as complex partial seizures. Other features you may see in these complex partial seizures include staring, oral automatisms, unspecified automatic behavior, involuntary motor movements and/or head turning.
Furthermore, many patients have subtle nighttime behavioral changes, such as stretching, rubbing, and turning resembling a nighttime awakening. However, electroencephalography (EEG) studies during these events show abnormal electrical seizure activity, indicating that nocturnal behavioral events are actually subtle nocturnal seizures or non-convulsive status epilepticus. Many of these patients experience their seizures only during sleep. They can have seemingly bizarre features as they originate from the frontal lobe of the brain. Often, individuals with ring chromosome 20 syndrome are initially found to have complex partial seizures of frontal lobe origin, though imaging studies do not show a corresponding structural brain abnormality. In certain patients, these seizures may secondarily generalized.
Individuals from the ages of 0–17 years should be considered for ring 20 chromosome analysis if they have: predominantly complex partial seizures, medically refractory cryptogenic epilepsy, Lennox-Gastaut-like features with no cause identified, frequent subtle nocturnal seizures, an EEG showing prolonged high voltage frontally dominant slowing intermixed with spikes or sharp waves, an EEG showing overlapping features of continuous slow spike and wave discharges in sleep (CSWS) and electrical status epilepticus in sleep (ESES), and/or subsequent cognitive impairment/learning difficulties/mild retardation.These patients will typically have a normal childhood development until onset of epilepsy and lack evidence of dysmorphism or other congenital malformations.
Genetics
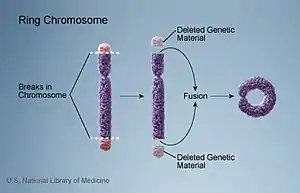
Rather than the typical linear pattern of a chromosome, deletion of the endings of a chromosome can lead to ring formation. A chromosome has two arms, one long and one short. Deletion of the short arm of chromosome 20 does not appear to result in epilepsy; however, terminal deletion of the long arm is associated with epilepsy. Therefore, some gene loss from the terminal segment could be responsible for the manifestation of epilepsy in ring chromosome 20 syndrome. The most common breakpoint in patients is in the q13.33 region of chromosome 20.
As chromosomes occur in pairs, the affected individual usually has one normal chromosome and the other replaced by a ring chromosome. However, this is not usually the case for every cell in the patient. In most individuals with ring chromosome 20 syndrome, a certain percentage have two normal chromosome 20s and the remainder have one normal and one ring 20 chromosome. This mixture is called mosaicism. A higher degree (percentage) of mosaicism appears to be associated with earlier age of seizure onset and presence of malformations. Yet, the degree of mosaicism does not determine response to drug treatment. Since the range in age of onset of seizures and IQ for a given mosaic ratio is relatively wide, prediction of phenotype based on the mosaicism ratio is somewhat limited.
Occasionally, there is a variation, where the ring chromosome 20 syndrome is characterized by an extra ring chromosome in addition to the two normal ones and hence gives rise to a partial trisomy or supernumerary ring 20. This supernumerary ring 20 can cause multiple anomalies but no epilepsy, and this resulting syndrome should be differentiated from ring chromosome 20 syndrome. This confusion has persisted even in the literature.
The genes in the q13.33 region of chromosome 20 have yet to be fully delineated. However, this telomeric region includes two genes that are related to other distinct epilepsy syndromes, autosomal dominant nocturnal frontal lobe epilepsy and benign familial neonatal convulsions. The genes identified in these two syndromes are the nicotinic acetylcholine receptor alpha-4 subunit and potassium voltage-gated channel subfamily KQT member 2 respectively. Mutations in these genes lead to the clinical manifestations found in these disorders. It is theorized that deletions in these regions from the ringed chromosome may lead to epilepsy in affected patient. A similar association between a ring chromosome and an autosomal-dominant disease has been reported for ring 17 chromosome and Miller-Dieker syndrome.
The recurrence risk for ring chromosome 20 syndrome is very low. The ring formation is typically not identified in parents, and occurs 'de novo' during pre-natal development.
Diagnosis
The ring 20 abnormality may be limited to as few as 5% of cells, so a screen for chromosomal mosaicism is critical. Newer array technology will not detect the ring chromosome and the standard metaphase chromosome analysis has been recommended. A karyotype analysis examining at least 50 cells should be requested to properly detect mosaicism.
Treatment
Successful management of seizures plays a key role in improving quality of life. Antiepileptic medications are the main therapies for seizures; however, it appears that seizures in this syndrome do not respond well to drugs. In the cases reported in literature, numerous new and old antiepileptic drugs have been tried, but no one drug appears to be more efficacious than others. Therefore, no recommendations can be made regarding the selection of the most appropriate antiepileptic drug. As not all cases of ring chromosome 20 syndrome are the same, different individuals may respond to treatment in different ways.Alternates to antiepileptic drug treatment include the ketogenic diet and vagus nerve stimulation but not epilepsy surgery.
Ketogenic Diet
The ketogenic diet is a high fat, low carbohydrate diet reserved for intractable childhood epilepsies. There are no published reports on the use of the ketogenic diet in patients with ring chromosome 20 syndrome. However, its efficacy and safety are well established in other difficult to control epilepsy syndromes.
Epilepsy Surgery
Epilepsy surgery investigations are performed to identify a discrete seizure focus. Extensive investigations in ring chromosome 20 syndrome patients fails to identify a discrete seizure focus and published data supports that that epilepsy in ring chromosome 20 syndrome is not amenable to resective surgery. The Cost ranges between £50,000 - £200,000.
VNS Therapy
The vagal nerve stimulator is a battery-powered device similar to a pacemaker that is implanted under the skin. It delivers a mild electrical stimulation to the brain via the vagus nerve and has proven to be effective for the treatment of complex partial seizures. There are only a few published reports on the success of this therapy in ring chromosome 20 epilepsy syndrome, making it unclear if this is the optimal therapy.
Prognosis
Limited data is available for the long-term prognosis of ring chromosome 20 syndrome since only over 60 patients with this syndrome have been reported in published literature. Optimal control of seizures appears to be the determining factor, but early diagnosis and a comprehensive management plan with multidisciplinary support is also thought be to be important.
Research
Two international research studies are currently underway. The International Genetic Study done with the Spinner Laboratory at The Children's Hospital of Philadelphia studies the ring 20 chromosome at the molecular level. The Clinical Research Study collects clinical information from parents to create a database of about the full spectrum of patients with ring chromosome 20 syndrome.
References
- Reference, Genetics Home. "ring chromosome 20 syndrome". Genetics Home Reference. Retrieved 2017-03-18.
- "Ring chromosome 20 - Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". Rarediseases.info.nih.gov. Retrieved 1 December 2017.
- "Orphanet: Ring chromosome 20 syndrome". Orpha.net. Retrieved 1 December 2017.
Further reading
- Schinzel A, Niedrist D (2001). "Chromosome imbalances associated with epilepsy". Am. J. Med. Genet. 106 (2): 119–24. doi:10.1002/ajmg.1576. PMID 11579431.