XX male syndrome
XX male syndrome, also known as de la Chapelle syndrome, is a rare congenital intersex condition in which an individual with a 46, XX karyotype (otherwise associated with females) has phenotypically male characteristics that can vary among cases.[2] Synonyms include 46,XX testicular difference of sex development (46,XX DSD), 46,XX sex reversal, nonsyndromic 46,XX testicular DSD, and XX sex reversal.[3][4][5][6]
| XX male syndrome | |
|---|---|
| Other names | De la Chapelle syndrome[1] |
_Karyotype_Human_46%252CXX_(woman).jpg.webp) | |
| Human karyotype 46 XX | |
| Specialty | Medical genetics |
In 90 percent of these individuals, the syndrome is caused by the Y chromosome's SRY gene, which triggers male reproductive development, being atypically included in the crossing over of genetic information that takes place between the pseudoautosomal regions of the X and Y chromosomes during meiosis in the father.[2][7] When the X with the SRY gene combines with a normal X from the mother during fertilization, the result is an XX male. Less common are SRY-negative XX males, which can be caused by a mutation in an autosomal or X chromosomal gene.[2] The masculinization of XX males is variable.
This syndrome is diagnosed through various detection methods and occurs in approximately 1:20,000 newborn males, making it much less common than Klinefelter syndrome.[2][8][9] Treatment is medically unnecessary, although some individuals choose to undergo treatments to make them appear more male or female.[1][10] The alternative name for XX male syndrome refers to Finnish scientist Albert de la Chapelle, who studied the condition and its etiology.[11]
Signs and symptoms
The appearance of XX males can fall into one of three categories: 1) males that have normal internal and external genitalia, 2) males with external ambiguities, and 3) males that have both internal and external genital ambiguities.[12] External genital ambiguities can include hypospadias, micropenis, and clitoromegaly.[12] Typically, the appearance of XX males differs from that of an XY male in that they are smaller in height and weight.[2] Most XX males have small testes, and have an increase in maldescended testicles compared to XY males. Many are sterile.[2][13] Some XX male individuals have decreased amounts of body hair and decreased libido.[13] Individuals with this condition sometimes have feminine characteristics, with varying degrees of gynecomastia but with no intra-abdominal Müllerian tissue.[13] According to research at the University of Oklahoma health science centers, despite XX males exhibiting feminine characteristics, their behaviours are usually representative of masculinity in their culture.[14]
They generally have small testes and may also have abnormalities such as undescended testes (cryptorchidism) or the urethra opening on the underside of the penis (hypospadias). A small number of affected people have external genitalia that do not look clearly male or clearly female (ambiguous genitalia). Affected children are typically raised as males and are likely to have a male gender identity.[3]
Masculinization
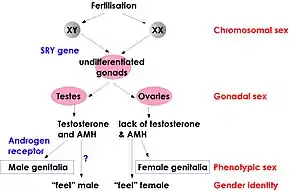
The degree to which individuals with XX male syndrome develop the male phenotype is variable, even among SRY-positive individuals.[15] A completely male phenotype usually develops in the presence of the SRY gene but, in some cases, the presence of the SRY gene can result in internal and/or external genitalia ambiguities.[15] Normal XX females undergo X inactivation during which one copy of the X chromosome is silenced. It is thought that X inactivation in XX males may account for the genital ambiguities and incomplete masculinization seen in SRY-positive XX males.[16][15] The X chromosome with the SRY gene is preferentially chosen to be the active X chromosome 90% of the time, which explains complete male phenotype being observed often in SRY-positive XX males.[16][15] In the remaining 10%, X inactivation spreads to include a portion of the SRY gene, resulting in incomplete masculinization.[16][15]
Masculinization of SRY-negative XX males is dependent upon which genes have mutations and at what point in development these mutations occur.[17]
Genetics
Males typically have one X chromosome and one Y chromosome in each diploid cell of their bodies. Females typically have two X chromosomes. XX males that are SRY-positive have two X chromosomes, with one of them containing genetic material (the SRY gene) from the Y chromosome; this gene causes them to develop a male phenotype despite having chromosomes more typical of females.[2] Some XX males, however, do not have the SRY gene (SRY-negative) and the male phenotype may be caused by another gene on one of the autosomes.
SRY-positive
The SRY gene, normally found on the Y chromosome, plays an important role in sex determination by initiating testicular development. In about 80 percent of XX males, the SRY gene is present on one of the X chromosomes.
The condition results from an abnormal exchange of genetic material between chromosomes (translocation). This exchange occurs as a random event during the formation of sperm cells in the affected person's father. The tip of the Y chromosome contains the SRY gene and, during recombination, a translocation occurs in which the SRY gene becomes part of the X chromosome.[12][18] If a fetus is conceived from a sperm cell with an X chromosome bearing the SRY gene, it will develop as a male despite not having a Y chromosome. This form of the condition is called SRY-positive 46,XX testicular disorder of sex development.[3]
SRY-negative
About 20 percent of those with 46 XX testicular disorder of sex development do not have the SRY gene. This form of the condition is called SRY-negative 46,XX testicular disorder of sex development. The cause of the disorder in these individuals is often unknown, although changes affecting other genes have been identified. Individuals with SRY-negative 46,XX testicular disorder of sex development are more likely to have ambiguous genitalia than are people with the SRY-positive form.[3][2]
The exact cause of this condition is unknown but it has been proposed that mutations in the SOX9 gene may contribute to this syndrome since SOX9 plays a role in testes differentiation during development.[19][17] Another proposed cause is mutations to the DAX1 gene, which encodes a nuclear hormone receptor.[20][21] DAX1 represses masculinizing genes; therefore, if there is a loss of function of DAX1, then testes can develop in an XX individual.[21] Mutations in SF1 and WNT4 genes have also been studied in connection with SRY-negative XX male syndrome.[21]
Diagnosis
In cases where the individual is being evaluated for ambiguous genitalia, such as a small phallus, hypospadias, or labioscrotal folds, exploratory surgery may be used to determine if male and/or female internal genitalia is present.[22] Indicators include two testes which have not descended the inguinal canal, although this is seen in a minority of XX males, and the absence of Müllerian tissue.[13] External indicators include decreased body weight and small testes.[2]
A standard karyotype can be completed to cytogenetically determine that an individual with a partial or complete male phenotype has an XX genotype.[12][22][23]
The presence and location of the SRY gene can by determined using fluorescence in situ hybridization (FISH).[2][15]
Treatment
Genital ambiguities, while not necessary to treat for medical reasons, can be treated with hormonal therapy, surgery, or both. Since XX male syndrome is variable in its presentation, the specifics of treatment varies widely as well. In some cases, gonadal surgery can be performed to remove partial or whole female genitalia. This may be followed by plastic and reconstructive surgery to make the individual appear more externally male.[24] Conversely, the individual may wish to become more feminine and feminizing genitoplasty can be performed to make the ambiguous genitalia appear more female.[25] Hormonal therapy may also aid in making an individual appear more male or female.[24][25]
Testosterone

At puberty, most affected individuals require treatment with the male sex hormone testosterone to induce development of male secondary sex characteristics such as facial hair and deepening of the voice (masculinization). Hormone treatment can also help prevent breast enlargement (gynecomastia). Adults with this disorder are usually shorter than average for males and are unable to have children (infertile).[3]
Epidemiology
As of 2010, only 200 cases have been reported — it is estimated that 1 of every 20,000 to 30,000 males has a 46,XX karyotype.[26][27][3]
See also
- X chromosome, for other conditions related to the X chromosome
- For a condition that causes people who have XY chromosomes to have an ambiguous or feminine phenotype, see androgen insensitivity syndrome (AIS)
- For a second condition that causes people who have XY chromosomes to have a feminine phenotype, see XY gonadal dysgenesis (also known as Swyer syndrome)
- Karyotype
- Disorders of sex development
- Intersex medical interventions
References
- de la Chapelle, A (January 1972). "Analytic review: nature and origin of males with XX sex chromosomes". American Journal of Human Genetics. 24 (1): 71–105. ISSN 0002-9297. PMC 1762158. PMID 4622299.
- Vorona, Elena; Zitzmann, Michael; Gromoll, Jörg; Schüring, Andreas N.; Nieschlag, Eberhard (2007-09-01). "Clinical, Endocrinological, and Epigenetic Features of the 46,XX Male Syndrome, Compared with 47,XXY Klinefelter Patients". The Journal of Clinical Endocrinology & Metabolism. 92 (9): 3458–3465. doi:10.1210/jc.2007-0447. ISSN 0021-972X. PMID 17579198.
- "46,XX testicular disorder of sex development - Genetics Home Reference". Archived from the original on 2019-05-17. Retrieved 2017-01-08.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: 46,XX testicular disorder of sex development". www.orpha.net. Archived from the original on 2017-01-13. Retrieved 2017-01-12.
- Délot, EC; Vilain, EJ; Pagon, RA; Adam, MP; Ardinger, HH; Wallace, SE; Amemiya, A; Bean, LJH; Bird, TD; Fong, CT; Mefford, HC; Smith, RJH; Stephens, K (1993). "Nonsyndromic 46,XX Testicular Disorders/Differences of Sex Development". Nonsyndromic 46,XX Testicular Disorders of Sex Development. GeneReviews. University of Washington, Seattle. PMID 20301589. Archived from the original on 18 January 2017. Retrieved 12 January 2017.updated 2015
- "46,XX testicular disorder of sex development: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 2020-09-15. Retrieved 2020-09-06.
- Andersson, M.; Page, D. C.; de la Chapelle, A. (1986-08-15). "Chromosome Y-specific DNA is transferred to the short arm of X chromosome in human XX males". Science. 233 (4765): 786–788. Bibcode:1986Sci...233..786A. doi:10.1126/science.3738510. ISSN 0036-8075. PMID 3738510. S2CID 32456133.
- Anık, Ahmet; Çatlı, Gönül; Abacı, Ayhan; Böber, Ece (2013). "46,XX Male Disorder of Sexual Development: A Case Report". Journal of Clinical Research in Pediatric Endocrinology. 5 (4): 258–260. doi:10.4274/Jcrpe.1098. ISSN 1308-5727. PMC 3890225. PMID 24379036.
- Ucan, Bekir; Ozbek, Mustafa; Topaloglu, Oya; Yesilurt, Ahmet; Gungunes, Askin; Demrici, Taner; Delibasi, Tunfay (July 2012). "46,XX Male Syndrome". Turkish Journal of Endocrinology and Metabolism. 17 (2): 46–48. doi:10.4274/Tjem.2064.
- de la Chapelle, Albert (1985). Cytogenetics of the mammalian X-chromosome, Part B: Progress and topics in cytogenetics. New York: Alan Liss. pp. 75–85.
- de la Chapelle, Albert (August 1981). "The etiology of maleness in XX men". Human Genetics. 58 (1): 105–116. doi:10.1007/bf00284157. ISSN 0340-6717. PMID 6945286. S2CID 26425178.
- Chen, Harold (2012). "XX Male". Atlas of Genetic Diagnosis and Counseling. pp. 2191–2196. doi:10.1007/978-1-4614-1037-9_250. ISBN 978-1-4614-1036-2.
- Lisker, R; Flores, F; Cobo, A; Rojas, F G (December 1970). "A case of XX male syndrome". Journal of Medical Genetics. 7 (4): 394–398. doi:10.1136/jmg.7.4.394. ISSN 0022-2593. PMC 1468937. PMID 5501706.
- Abusheikha, N.; Lass, A.; Brinsden, P. (2001-04-01). "XX males without SRY gene and with infertility: Case report". Human Reproduction. 16 (4): 717–718. doi:10.1093/humrep/16.4.717. ISSN 0268-1161. PMID 11278224.
- Kusz, Kamila; Kotecki, Maciej; Wojda, Alina; Szarras-Czapnik, Maria; Latos-Bielenska, Anna; Warenik-Szymankiewicz, Alina; Ruszczynska-Wolska, Anna; Jaruzelska, Jadwiga (1999-06-01). "Incomplete masculinisation of XX subjects carrying the SRY gene on an inactive X chromosome". Journal of Medical Genetics. 36 (6): 452–456. doi:10.1136/jmg.36.6.452. ISSN 0022-2593. PMC 1734388. PMID 10874632.
- Bouayed Abdelmoula, Nouha; Portnoi, Marie-France; Keskes, Leila; Recan, Dominique; Bahloul, Ali; Boudawara, Tahia; Saad, Ali; Rebai, Tarek (2003-01-01). "Skewed X-chromosome inactivation pattern in SRY positive XX maleness: a case report and review of literature". Annales de Génétique. 46 (1): 11–18. doi:10.1016/S0003-3995(03)00011-X. PMID 12818524.
- Rajender, S. (2006-05-01). "SRY-negative 46,XX male with normal genitals, complete masculinization and infertility". Molecular Human Reproduction. 12 (5): 341–346. doi:10.1093/molehr/gal030. ISSN 1360-9947. PMID 16556678.
- Margarit, Ester; Coll, M. Dolors; Oliva, Rafael; Gómez, David; Soler, Anna; Ballesta, Francisca (2000-01-03). "SRY gene transferred to the long arm of the X chromosome in a Y-positive XX true hermaphrodite". American Journal of Medical Genetics. 90 (1): 25–28. doi:10.1002/(sici)1096-8628(20000103)90:1<25::aid-ajmg5>3.0.co;2-5. ISSN 1096-8628. PMID 10602113.
- Vetro, Annalisa; Ciccone, Roberto; Giorda, Roberto; Patricelli, Maria Grazia; Mina, Erika Della; Forlino, Antonella; Zuffardi, Orsetta (2011-01-01). "XX males SRY negative: a confirmed cause of infertility". Journal of Medical Genetics. 48 (10): jmedgenet–2011–100036. doi:10.1136/jmedgenet-2011-100036. ISSN 0022-2593. PMC 3178810. PMID 21653197.
- Swain, Amanda; Narvaez, Veronica; Burgoyne, Paul; Camerino, Giovanna; Lovell-Badge, Robin (1998-02-19). "Dax1 antagonizes Sry action in mammalian sex determination". Nature. 391 (6669): 761–767. Bibcode:1998Natur.391..761S. doi:10.1038/35799. ISSN 1476-4687. PMID 9486644. S2CID 4416667.
- Domenice, S.; Corrêa, R. V.; Costa, E. M. F.; Nishi, M. Y.; Vilain, E.; Arnhold, I. J. P.; Mendonca, B. B. (January 2004). "Mutations in the SRY, DAX1, SF1 and WNT4 genes in Brazilian sex-reversed patients". Brazilian Journal of Medical and Biological Research. 37 (1): 145–150. doi:10.1590/S0100-879X2004000100020. ISSN 0100-879X. PMID 14689056.
- "OMIM Entry - # 400045 - 46,XX SEX REVERSAL 1; SRXX1". www.omim.org. Archived from the original on 2019-12-13. Retrieved 2017-11-07.
- Délot, Emmanuèle C; Vilain, Eric J (2003). "Nonsyndromic 46,XX Testicular Disorders/Differences of Sex Development". Nonsyndromic 46,XX Testicular Disorders of Sex Development. GeneReviews. University of Washington, Seattle. PMID 20301589. Archived from the original on 23 June 2020. Retrieved 6 December 2018.
- Parada-Bustamante, Alexis; Ríos, Rafael; Ebensperger, Mauricio; Lardone, María Cecilia; Piottante, Antonio; Castro, Andrea (2010-11-01). "46,XX/SRY-negative true hermaphrodite". Fertility and Sterility. 94 (6): 2330.e13–2330.e16. doi:10.1016/j.fertnstert.2010.03.066. ISSN 0015-0282. PMID 20451191. Archived from the original on 2019-12-13. Retrieved 2017-11-29.
- Kurita, Masakazu; Aiba, Emiko; Matsumoto, Daisuke; Sato, Katsujiro; Nagase, Takashi; Yoshimura, Kotaro (May 2006). "Feminizing genitoplasty for treatment of XX male with masculine genitalia". Plastic and Reconstructive Surgery. 117 (6): 107e–111e. doi:10.1097/01.prs.0000214653.30135.a1. ISSN 1529-4242. PMID 16651931.
- Bouvattier, Claire (1 January 2010). "Disorders of Sex Development". Pediatric Urology (Second Edition). W.B. Saunders: 459–475. doi:10.1016/B978-1-4160-3204-5.00035-9. ISBN 9781416032045.
- Witchel, Selma Feldman; Topaloglu, A. Kemal (1 January 2019). "Chapter 17 - Puberty: Gonadarche and Adrenarche". Yen and Jaffe's Reproductive Endocrinology (Eighth Edition). Content Repository Only!: 394–446.e16. doi:10.1016/B978-0-323-47912-7.00017-2. ISBN 9780323479127. S2CID 239351840. Archived from the original on 17 May 2019. Retrieved 21 March 2020.
Further reading
- "Ambiguous Genitalia. Uncertain genetalia information. Patient | Patient". Patient. Retrieved 12 January 2017.