Ácidos quirales de Lewis
Los ácidos quirales de Lewis (CLA) son un tipo de catalizador ácido de Lewis que afecta la quiralidad del sustrato cuando reacciona con él. En tales reacciones, la síntesis favorece la formación de un enantiómero o diastereómero específico. El método entonces es una reacción de síntesis asimétrica enantioselectiva. Dado que afectan la quiralidad, producen productos ópticamente activos a partir de materiales de partida ópticamente inactivos o mixtos. Este tipo de formación preferencial de un enantiómero o diastereómero sobre el otro se conoce formalmente como una inducción asimétrica. En este tipo de ácido de Lewis, el átomo aceptor de electrones es típicamente un metal, tal como indio, zinc, litio, aluminio, titanio o boro. Los ligandos de alteración quiral empleados para sintetizar estos ácidos con mayor frecuencia tienen múltiples sitios básicos de Lewis (a menudo una estructura de diol o dinitrógeno) que permiten la formación de una estructura de anillo que involucra al átomo de metal.[1][2]
| Ácidos y Bases | |
|---|---|
 | |
| Ácidos y Bases | |
| |
| Tipos de ácidos | |
| Tipos de bases | |
Los ácidos de Achiral Lewis se han utilizado durante décadas para promover la síntesis de mezclas racémicas en innumerables reacciones diferentes. A partir de la década de 1960, los químicos han utilizado los ácidos quirales para inducir las reacciones enantioselectivas. Los tipos de reacción comunes incluyen las reacciones de Diels-Alder, la reacción de ene, las reacciones de cicloadición [2+2], la hidrocianación de aldehídos y, más notablemente, las expoxidaciones de Sharpless.[3]
Teoría
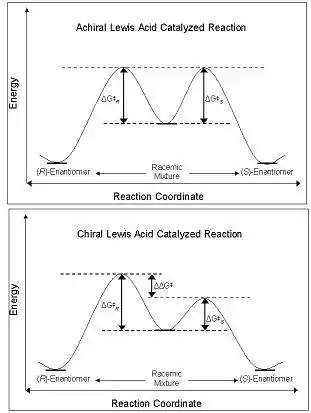
La enantioselectividad de los CLA se deriva de su capacidad para perturbar la barrera de energía libre a lo largo de la ruta de coordenadas de reacción que conduce al enantiómero R o S. Los diastereómeros y enantiómeros en estado fundamental son de igual energía en el estado fundamental, y cuando reaccionan con un ácido lewis aquiral, sus intermedios diastereoméricos, estados de transición y productos también tienen la misma energía. Esto conduce a la producción de mezclas racémicas de productos. Sin embargo, cuando se usa un CLA en la misma reacción, la barrera energética de la formación de un diastereómero es menor que la de otro: la reacción está bajo control cinético. Si la diferencia en las barreras de energía entre los estados de transición diastereoméricos es de magnitud suficiente, entonces debe observarse un alto exceso enantiomérico de un isómero (Figura 2).[4]
Aplicaciones de los CLAs en síntesis asimétrica
Reacción Diels-Alder
Las reacciones de Diels-Alder ocurren entre un dieno conjugado y un alqueno (comúnmente conocido como el dienófilo). Este proceso de cicloadición permite la formación estereoselectiva de anillos de ciclohexeno capaces de poseer hasta cuatro centros estereogénicos contiguos.
Las reacciones de Diels-Alder pueden conducir a la formación de una variedad de isómeros estructurales y estereoisómeros. La teoría de los orbitales moleculares considera que se favorece el estado de transición endo, en lugar del estado de transición exo (regla de adición endo). Además, las interacciones orbitales secundarias aumentadas se han postulado como la fuente de diastereoselección endo mejorada.
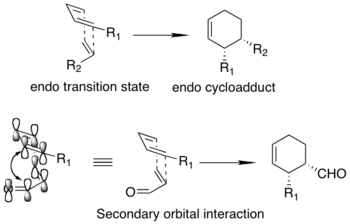
La adición de un CLA activa selectivamente un componente de la reacción (el dieno o el dienófilo) al tiempo que proporciona un entorno estereodefinido que permite una enantioselectividad única.
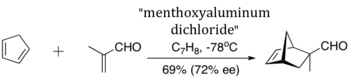
Koga y colaboradores describieron el primer ejemplo práctico de una reacción enantioselectiva catalítica de Diels-Alder promovida por un CLA (dicloruro de mentoxialuminio) derivado del mentol y el dicloruro de etilaluminio.[5]
Una década más tarde, Elias James Corey introdujo un efectivo controlador de diamina de aluminio para la reacción de Diels-Alder. La formación del catalizador activo se logra mediante el tratamiento de la bis (sulfonamida) con trimetilaluminio; la recuperación del ligando fue esencialmente cuantitativa. El tetracoordinado de aluminio propuesto evita que la imida actúe como una base de Lewis quelante, al tiempo que mejora el α-vinil protón del dienfilo y el protón bencílico del catalizador.
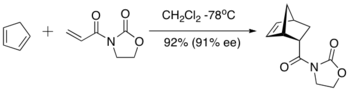
La estructura de rayos X del catalizador mostraba un ambiente estereodefinido.[6]
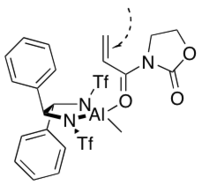
En 1993, Wulff y sus colegas encontraron que un complejo derivado del cloruro de dietilaluminio y un ligando de biarilo "abovedado" a continuación catalizaron la reacción enantioselectiva Diels-Alder entre ciclopentadieno y metacroleína. El ligando quiral se recupera cuantitativamente mediante cromatografía en gel de sílice.[7]
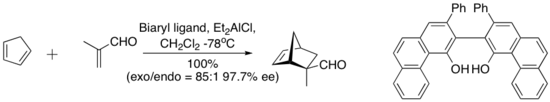
Hisashi Yamamoto y sus colaboradores han desarrollado un práctico catalizador Diels-Alder para dienófilos aldehídos. El complejo quiral (aciloxi) borano (CAB) es eficaz para catalizar varias reacciones de aldehído Diels-Alder. Los experimentos espectroscópicos de RMN indicaron la proximidad del aldehído y el anillo arilo. Además, el apilamiento de Pi entre el grupo arilo y el aldehído se sugirió como una característica organizativa que impartía una alta enantioselectividad a la cicloadición.[8]
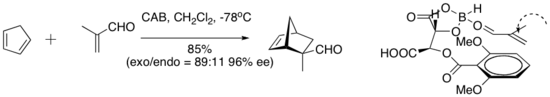
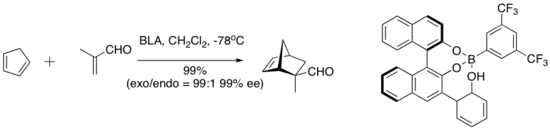
Yamamoto y sus compañeros de trabajo han introducido una serie de catalizadores conceptualmente interesantes que incorporan un protón ácido en el catalizador activo. Este tipo de lo que se denomina ácido de Lewis quiral asistido con ácido de Bronsted (BLA) cataliza una serie de reacciones de cicloadición de dieno-aldehído.[9]
Reacción aldólica
En la reacción aldólica, la diastereoselectividad del producto a menudo es dictada por la geometría del enolato de acuerdo con el modelo de Zimmerman-Traxler. El modelo predice que el enolato Z dará productos sin y que los enolatos E darán productos anti. Los ácidos de Lewis quiral permiten productos que desafían el modelo de Zimmerman-Traxler y permiten el control de la estereoquímica absoluta. Kobayashi y Horibe demostraron esto en la síntesis de derivados de dihidroxitioéster, utilizando un ácido de Lewis quiral a base de estaño.[10]
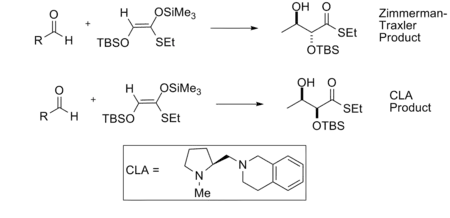
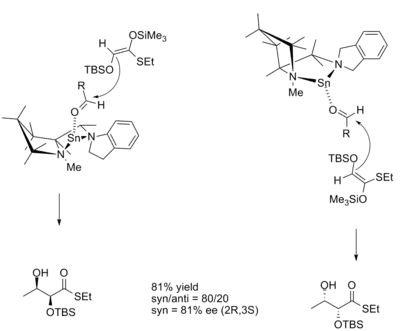
Las estructuras de transición para las reacciones con los enantiómeros catalizadores tanto R como S se muestran a continuación.
Reacción de Baylis-Hillman
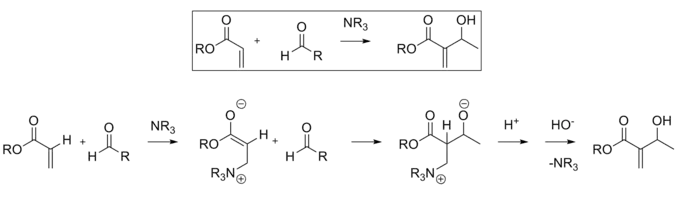
La reacción de Baylis-Hillman es una ruta para la formación de enlaces CC entre un carbonilo alfa, beta insaturado y un aldehído, que requiere un catalizador nucleófilo, generalmente una amina terciaria, para una adición y eliminación de tipo Michael. La estereoselectividad de estas reacciones suele ser pobre. Chen et al. demostró una reacción enantioselectiva quiral de Lewis catalizada por ácido. Se utilizó lantano en este caso. De manera similar, también se puede usar una amina quiral para lograr la estereoselectividad.[11]
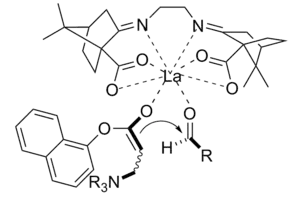
El producto obtenido por la reacción usando el catalizador quiral se obtuvo con buen rendimiento con excelente enantioselectividad.
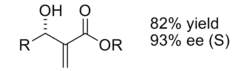
Reacción ene
Los ácidos lewis quirales también han demostrado ser útiles en la reacción de eno. Cuando es catalizada por un ácido lequítico aquiral, la reacción normalmente proporciona una buena diastereoselectividad.[12]
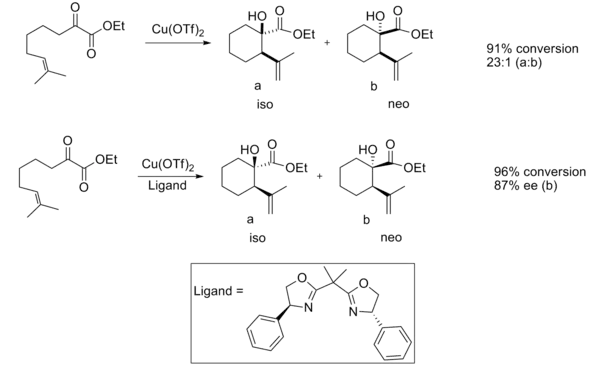
Cuando se usó un catalizador ácido lewis quiral se observó una buena enantioselectividad.
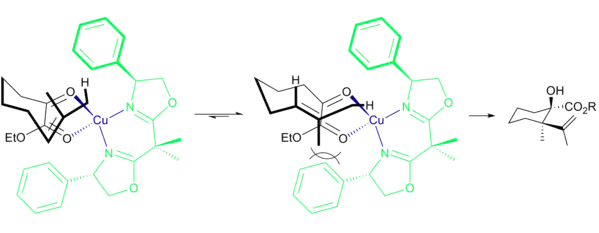
Se cree que la enantioselectividad se debe a las interacciones estéricas entre el grupo metilo y fenilo, lo que hace que la estructura de transición del producto iso sea considerablemente más favorable.
Ejemplos de ácidos de Lewis aquirales en síntesis estereoselectiva
Acoplamiento catalizado por níquel de 1,3-dienos con aldehídos En algunos casos, un ácido de Lewis aquiral puede proporcionar una buena estereoselectividad. Kimura et al. demostró el acoplamiento regio y diastereoselectivo de 1,3- dienos con aldehídos.[13]
Utilidad de los ácidos de Lewis quirales
La síntesis asimétrica y la producción de sustancias enantioméricamente puras mediante el uso de CLA son de particular interés para químicos orgánicos y corporaciones farmacéuticas. Debido a que muchos productos farmacéuticos se dirigen a enzimas que son específicas para un enantiómero particular, los compuestos destinados a la administración al paciente deben ser de una alta pureza óptica. Además, la resolución de un enantiómero particular a partir de una mezcla racémica es costosa y derrochadora.
Notas
- Lewis Acid Reagents. A Practical Approach. Yamamoto, H., Oxford University Press. 1999 (accessed December 3, 2008)
- Bin, Y., Pikul, S., Imwinkelried, R., Corey, E.J. 1989, JACS, (14) 5493-5495
- Narasaka, K. Synthesis. 1991 (01) 1-11
- Morrison, J.D., Mosher, H.S. (1971). Asymmetric Organic Reactions. Prentice-Hall, Inc. ISBN 978-0-13-049551-8.
- Hashimoto S-I, Komeshima N, Koga K, 1979, J Chem Soc Chem Commun, 437
- Coery, EJ; Sarshar, S; Bordner, J, 1992, J Am Chem Soc, 114, 7938
- Bao, J; Wulff, WD; Rheingold, AL, 1993, J Am Chem Soc, 115, 3814
- Ishihara, K; Gao, Q; Yamamoto, H, 1993, J Am Chem Soc, 115, 10412
- Ishihara, K; Yamamoto, H, 1994, J Am Chem Soc, 116, 1561
- Kobayashi, S.; Horibe, M., 1997, Chem. Eur. J., 3, 9, 1472-1481
- Yang, K.; Lee, W.; Pan, J.; Chen, K., 2003, J. Org. Chem., 68, 915-919
- Yang, D.; Yang, M.; Zhu, N., 2003 Org. Lett., 5, 20, 3749-3752
- Kimura, M.; Ezoe, A,; Mori, M.; Iwata, K.; Tamaru., Y., 2006, JACS, 128, 8559-8568
Referencias
- Reactivos de ácido de Lewis. Un enfoque práctico. Yamamoto, H., Oxford University Press., 1999
- Bin, Y., Pikul, S., Imwinkelried, R., Corey, EJ 1989, JACS, (14) 5493-5495
- Narasaka, K. 1991, Síntesis, (01) 1-11
- Reacciones orgánicas asimétricas. Morrison, JD, Mosher, HS Prentice-Hall, Inc., 1971
| Control de autoridades |
|
|---|
 Datos: Q5101822
Datos: Q5101822