Cicloadición 2+2
La cicloadición [2+2] es una reacción de cicloadición, que generalmente implica la formación de nuevas moléculas por la reacción de dos moléculas insaturadas a través de dos de los electrones de cada una de ellas (de ahí "[2+2]"). Normalmente se trata de una reacción fotoquímica, el uso de alguna forma de luz (generalmente denotado h·ν), en oposición a un proceso térmico.
Mecanismos de las cicloadiciones [2+2]
Hay 4 posibles mecanismos para la cicloadición [2 +2]: [π2s+π2s], [π2a+π2s], [π2s+π2a], [π2a+π2a]. Sin embargo, en la realidad, la única reacción que, en general, se puede producir debido a las limitaciones geométricas es la suprafacial con respecto a ambos componentes.[1]
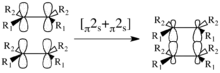
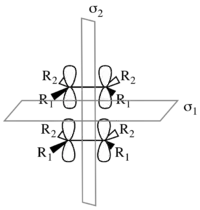
Considerando solo la cicloadición [π2s+π2s], el mecanismo conduce a una retención de la estereoquímica en el producto, como se ilustra a la derecha. Hay dos elementos de simetría presentes en los materiales de partida, el estado de transición y de productos: σ1 y σ2. σ1 es el plano de simetría entre los componentes perpendiculares a los orbitales p; σ2 divide las moléculas en un plano perpendicular a los enlaces-σ.[2]
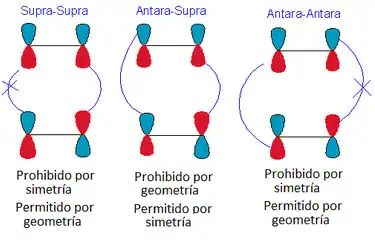
Una cicloadición [2 +2] es un proceso de 4 electrones que sigue las reglas de Woodward-Hoffmann. Estas reglas nos dicen que la aproximación suprafacial-suprafacial está prohibida por simetría en condiciones térmicas, pero permitida en condiciones fotoquímicas; la aproximación suprafacial-antarafacial está permitida por simetría, pero prohibida por la geometría; y la aproximación antarafacial-antarafacial está prohibida por la simetría en condiciones térmicas, pero permitida en condiciones fotoquímicas:
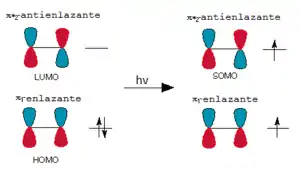
Los alquenos solo dan la cicloadición [2+2] por vía fotoquímica debido a que para que se de la misma, se tiene que promocionar un electrón del orbital HOMO al LUMO. Para que se produzca la excitación del electrón de la molécula del alqueno hace falta irradiar en la longitud de onda adecuada, y esta no es otra que la radiación ultravioleta o la luz visible. La promoción del electrón no cambia la geometría del alqueno, pero cambia la simetría del orbital, de tal manera que el orbital SOMO del alqueno excitado ahora ya puede reaccionar con el orbital LUMO del alqueno no excitado al no estar prohibida por simetría ni geometría esta reacción. Por via térmica se produce la cicloadición [2+2] de alenos y cetenas, porque poseen una geometría diferente al poseer dos dobles enlaces consecutivos.[1]
Cicloadiciones [2+2] fotoquímicas
Como ya se ha comentado anteriormente, los alquenos solo dan cicloadiciones [2+2] fotoquímicas debido a las reglas de selección impuestas por la química cuántica. Generalmente se producen por irradiación con luz ultravioleta (UV) entre 250-375 nm.
Debido al uso de luz UV suelen tratarse de reacciones no concertadas pese al hecho de ser una reacción pericíclica típica. Esto se debe a que la luz UV es lo suficientemente energética para producir radicales libres por medio de la fotodisociación del doble enlace. Todo esto, junto con las posibilidades de unión a la hora de formarse los nuevos enlaces σ, según la disposición geométrica en el estado de transición, hace que estas reacciones sean generalmente poco estereoselectivas.
Cicloadiciones [2+2] de alquenos
Un ejemplo clásico de cicloadición [2+2] fotoquímica de alquenos sería la dimerización fotoquímica del ácido cinámico, cuya reacción entre los dos alquenos produce la síntesis de distintos isómeros del ácido truxílico.[3] Los dos alquenos trans reaccionan cabeza-con-cola, y el isómero aislado es denominado ácido truxílico.
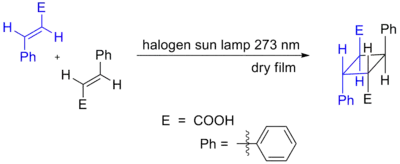 Cinnamic Acid CycloAddition
Cinnamic Acid CycloAddition
Estas reacciones al no estar orientadas por ningún grupo director, suele ser interesante usar algún compuesto químico auxiliar que ayude a que absorba la luz para la promoción del electrón del orbital HOMO al LUMO. A este compuesto auxiliar se le suele denominar fotosensibilizador, y provoca la promoción del electrón entre los orbitales frontera por un fenómeno físico de transferencia de energía. Generalmente se trata de un complejo inorgánico con un metal de transición. A continuación se pone un ejemplo de cicloadición [2+2] con un catalizador que produce la fotosensibilización:[4]
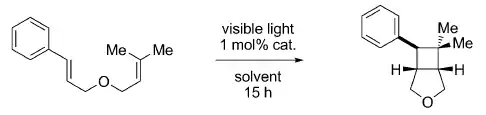
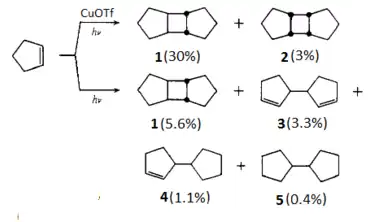
Es bien conocido el uso de sales de cobre (I), sobre todo del triflato de cobre (I) (CuOTf), como catalizador (o fotosensibilizador) en las cicloadiciones [2+2]. Se ha descrito el uso de estas sales para la fotodimerización de diferentes alquenos, entre ellos el ciclopenteno:[5]
Cicloadiciones [2+2] de alquenos con grupos electroatractores
La cicloadición [2+2] puede llevarse a cabo de manera no concertada a través de un intermedio zwitteriónico al hacer reaccionar alquenos con grupos ricos en densidad electrónica (enamina, vinil éteres...) con alquenos con grupos electrófilos (nitro, cianoetileno...):
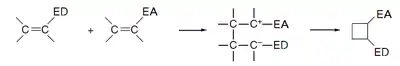
Algún ejemplo que se encuentra en la bibliografía sería:[6][7]
![Cicloadición [2+2] entre enamina y nitroalqueno](../I/Enamina_%252B_nitroalqueno.png.webp)
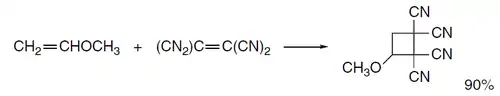
La estereoquímica de estas reacciones depende de la vida media del producto intermedio dipolar, que, a su vez, está influenciada por la polaridad del disolvente. En las reacciones de los éteres de enol con tetracianoetileno, la estereoquímica de la porción de éter de vinilo se mantiene en disolventes no polares. Sin embargo, en disolventes polares, la cicloadición no es estereoespecífica, debido a una mayor vida útil del intermedio zwitteriónico.[7]
Cicloadiciones [2+2] de enona-alqueno
El mecanismo de fotocicloadición [2 +2] se propone que empiece por la fotoexcitación de la enona a un estado excitado singlete. El estado singlete suele ser de muy corta duración, y decae a un estado triplete. En este punto, la enona forma un excímero con el alqueno en estado fundamental, con el tiempo dando un birradical triplete. Cuando cambia la inversión a singlete birradical permite el cierre del ciclobutano.[8] Como alternativa se propone un mecanismo de reacción pericíclica, en el que después de decaer al estado triplete, se forman un catión radical y un anión radical, que luego se recombinan para dar el ciclobutano.[9]
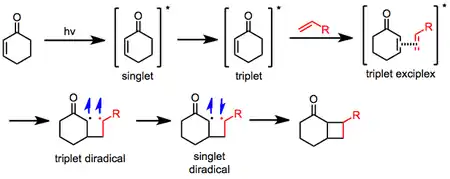
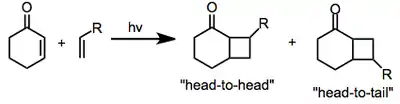
La fotocicloadición [2 +2] puede producir dos isómeros, dependiendo de la orientación de los sustituyentes en el alqueno y el grupo carbonilo de la enona. Cuando el carbonilo de la enona y el sustituyente de mayor prioridad están cerca, el isómero se denomina "cabeza a cabeza" (del inglés "head-to-head". Cuando el carbonilo de la enona y el sustituyente están lejos, el isómero se denomina "cabeza-a-cola" (del inglés "head-to-tail". La selectividad para uno de estos isómeros depende tanto de factores estéricos como electrónicos (ver más abajo).
La regioselectividad de la reacción está controlada principalmente por dos factores: las interacciones estéricas y las interacciones electrostáticas entre la enona excitada y el alqueno. En su estado excitado, la polaridad de las enonas se invierte de modo que el carbono β posee una carga parcial negativa. En el estado de transición para la formación del primer enlace, el alqueno tiende a alinearse de manera que el extremo negativo de su dipolo quede alejado del carbono β de la enona:[10]
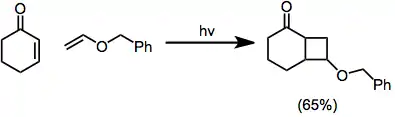
El impedimento estérico fomenta la colocación de grandes sustituyentes en lados opuestos del nuevo anillo de ciclobutano que se forma en la reacción de cicloadición:[10]
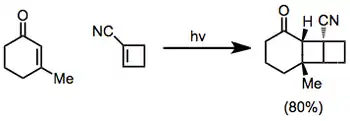
Las cicloadiciones [2 +2] intramoleculares pueden dar tanto productos "doblados" (del inglés "bent") como productos "derechos" (del inglés "straight")en función de la regioselectividad de la reacción. Cuando el fragmento conector entre la enona y el alqueno es de dos átomos de largo, los productos "doblados" predominan debido a la rápida formación de los anillos de cinco miembros.[11] Fragmentos conectores más largos tienden a dar productos "derechos".[12]
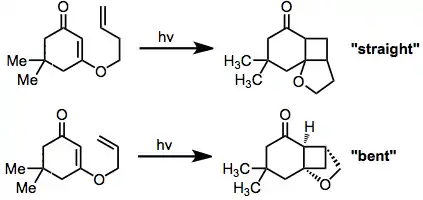
El fragmento conector también puede estar unido en la posición 2 de la enona en lugar de la 3 como se ha comentado anteriormente. Cuando el alqueno está unido aquí, los sustituyentes voluminosos en la posición 4 de la enona ejercen una diastereoselectividad moderada. En el siguiente ejemplo se ve cómo se forma el producto "derecho" pero tiene una mezcla de diastereoisómeros en función de si el grupo sustituyente en 4 está en "cis" o "trans" con respecto al ciclopentano del compuesto bicíclico:[13]

Cicloadiciones [2+2] térmicas
La cicloadición [2+2] térmica puede ir de tres formas: concertada, por vía radical o por vía iónica. Hay pocos ejemplos de cicloadiciones concertadas térmicas, donde se puede lograr la simetría orbital suprafacial-antarafacial para el cierre del anillo ([π2s+π2a]). Estas excepciones son las cicloadiciones [2+2] de cetenas.
Cicloadiciones [2+2] de cetenas
Las cicloadiciones de cetenas siguen un mecanismo concertado de cicloadición [2 +2]. Las cetenas, a diferencia de la mayoría de los alquenos, pueden alinearse antarafacialmente con respecto a otros alquenos debido a que posee dos dobles enlaces acumulados. Por lo tanto, la geometría suprafacial-antarafacial requerido para la vía concertada térmica de la cicloadición [2 +2] se puede lograr en las reacciones de cetenas.[14] Esta geometría tiene la consecuencia interesante que el sustituyente más voluminoso en la cetena tenderá a terminar en la cara del anillo de ciclobutanona más estéricamente impedida. En el estado de transición para la cicloadición, los sustituyentes pequeños apuntarán hacia el alqueno. Este modelo explica también la mayor reactividad de alquenos cis que trans en las cicloadiciones [2 +2] de cetenas.[15]
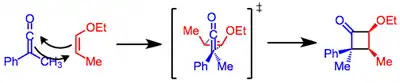
La configuración estereoquímica de la olefina se conserva en el producto de cicloadición. Los sustituyentes electroatractores en la cetena y los electrodadores en el alqueno aceleran la reacción,[16] pero las cetenas disustituidas reaccionan lentamente debido al impedimento estérico.[17]
La cetenas pueden dimerizar, o dos cetenas pueden reaccionar una con la otra para proporcionar ciclobutanonas sustituidas. En general, existen dos productos posibles en función de los dobles enlaces precisos que reaccionan:[18]

Las cetenas disustituidas dan sólo como producto la 1,3-ciclobutanodiona debido al impedimento estérico de los 2 sustituyentes de cada cetena.
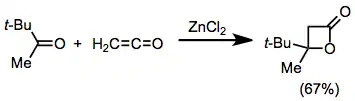
Además, reaccionan con aldehídos y cetonas para dar β-lactonas. Es necesario usar un ácido de Lewis para que catalice el proceso, a no ser que el carbonilo posea algún grupo buen electroatractor como sustutuyente:[19]
Referencias
- Woodward, R. B.; Hoffmann, Roald (1969). «The Conservation of Orbital Symmetry». Angew. Chem. Internat. Edit. 8 (11): 781-853. doi:10.1002/anie.196907811.
- Hoffmann, Roald; Woodward, R. B. (1965). «Selection Rules for Concerted Cycloaddition Reactions». J. Am. Chem. Soc. 87 (9): 2046. doi:10.1021/ja01087a034.
- Hein, Sara M. (junio de 2006). «An Exploration of a Photochemical Pericyclic Reaction Using NMR Data». Journal of Chemical Education 83: 940 - 942. doi:10.1021/ed083p940.
- Zhan Lu and Tehshik P. Yoon (2012). «Visible Light Photocatalysis of [2+2] Styrene Cycloadditions by Energy Transfer». Angew. Chem. 51: 10329 -10332. doi:10.1002/anie.201204835.
- Robert G. Salomon, Kirsten Folting, William E. Streib, and Jay K. Kochi (1974). «Copper (I) Catalysis in Photocycloadditions. II. Cyclopentene, Cyclohexene, and Cycloheptene». JACS 96: 1145-1152. doi:10.1021/ja00811a031.
- Martin E. Kuehne , Louise Foley (1965). «Reaction of Enamines with Nitro Olefins». J. Org. Chem. 30 (12): 4280-4284. doi:10.1021/jo01023a066.
- J. K. Williams , D. W. Wiley , B. C. McKusick (1962). «Cyanocarbon Chemistry. XIX.1,2 Tetracyanocyclobutanes from Tetracyanoethylene and Electron-rich Alkenes». J. Am. Chem. Soc. 84 (11): 2210-2215. doi:10.1021/ja00870a037.
- Loutfy, R. O.; DeMayo, P. (1972). «Primary Bond Formation in the Addition of Cyclopentenone to Chloroethylenes». Can. J. Chem. 50 (21): 3465. doi:10.1139/v72-560.
- Schmeling, N.; Hunger, K.; Engler, G.; Breiten, B.; Roelling, P.; (Mixa, A.; Staudt, C.; Kleinermanns, K. (2009). «Photo-crosslinking of poly[ethene-stat-(methacrylic acid)] functionalised with maleimide side groups». Polym. Int. 58 (7): 720. doi:10.1002/pi.2583.
- Corey, E. J.; Bass, J. D.; LeMahieu, R.; Mitra, R. B. (1964). J. Am. Chem. Soc. 86 (24): 5570. doi:10.1021/ja01078a034.
- Tamura, Y.; Kita, Y.; Ishibashi, H.; Ikeda, M. (1971). «Intramolecular photocycloaddition of 3-allyloxy- and 3-allylamino-cyclohex-2-enones: formation of oxa- and aza-bicyclo[2,1,1]hexanes». J. Chem. Soc. D. (19): 1167. doi:10.1039/C29710001167.
- Coates, R. M.; Senter, P. D.; Baker, W. R. (1982). «Annelative ring expansion via intramolecular [2 + 2] photocycloaddition of .alpha.,.beta.-unsaturated .gamma.-lactones and reductive cleavage: synthesis of hydrocyclopentacyclooctene-5-carboxylates». J. Org. Chem. 47 (19): 3597-3607. doi:10.1021/jo00140a001.
- Becker, D.; Haddad, N. (1986). «About the stereochemistry of intramolecular [2+2] photocycloadditions». Tetrahedron Lett. 27 (52): 6393. doi:10.1016/S0040-4039(00)87817-X.
- Moore, H. W.; Wilbur, D. S. (1980). «Cyanoketenes. Mechanism of tert-butylcyanoketene cycloaddition to aldo- and ketoketenes». J. Org. Chem. 45 (22): 4483-4491. doi:10.1021/jo01310a042.
- Rey, M.; Roberts, S.; Dieffenbacher, A.; Dreiding, A. S. Helv. Chim. Acta 1970, 53, 417.
- N. S. Isaacs; P. F. Stanbury (1970). «Steric factors in (2 + 2)-cycloadditions of ketens to olefins». J. Chem. Soc. D (17): 1969-1971. doi:10.1039/C29700001061.
- Huisgen, R.; Mayr, H. (1975). «Kinetics of 2+2 cycloadditions of diphenylketene to enol ethers; The structure of the orientation complexes». Tetrahedron Letters 16 (34): 2965-2968. doi:10.1016/S0040-4039(00)75045-3.
- Tenud, L.; Weilenmann, M.; Dallwigk, E. Helv. Chim. Acta 1977, 60, 975.
- Metzger, C.; Borrmann, D.; Wegler, R. Chem. Ber. 1967, 100, 1817.
| Control de autoridades |
|
|---|
 Datos: Q146615
Datos: Q146615