Ubiquinol-citocromo-c reductasa
El complejo III, citocromo bc1, coenzima Q - citocromo c reductasa o ubiquinol-citocromo-c reductasa es el tercer complejo de la cadena de transporte de electrones, que interviene en la respiración celular y la generación bioquímica de adenosín trifosfato (ATP) mediante fosforilación oxidativa. Estructuralmente, es una lipoproteína multimérica transmembrana, codificada tanto por el genoma nuclear como por el mitocondrial (concretamente, este último almacena la secuencia del citocromo b). Se encuentra en muchas bacterias y en las mitocondrias de los eucariotas aeróbicos.[2]
| Ubiquinol-citocromo-c reductasa | ||||
|---|---|---|---|---|
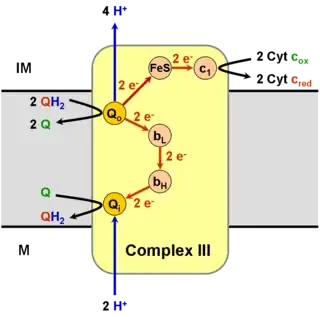 Esquema de las reacciones químicas que tienen lugar en el complejo III. | ||||
| Estructuras disponibles | ||||
| PDB | ||||
| Identificadores | ||||
| Identificadores externos |
Bases de datos de enzimas
| |||
| Número EC | 1.10.2.2 | |||
| Número CAS | 9027-03-6 | |||
| Ortólogos | ||||
| Especies |
| |||
| PubMed (Búsqueda) |
| |||
| PMC (Búsqueda) |
| |||
| Ubiquinol-citocromo-c reductasa | ||
|---|---|---|
 Estructura cristalina del complejo bc1 mitocondrial unido a la ubiquinona.[1] | ||
| Identificadores | ||
| Símbolo | UCR_TM | |
| Pfam | PF02921 | |
| InterPro | IPR004192 | |
| SCOP | 1be3 | |
| TCDB | 3.D.3 | |
| Familia OPM | 345 | |
| Proteína OPM | 3cx5 | |
El complejo III de consta de 11 subunidades y posee una masa de 248 kDa.[3] Tres de estas subunidades tienen función respiratoria (citocromo B, citocromo C1, proteína de Rieske) y poseen grupos prostéticos. La subunidad citocromo b alberga dos grupos hemo tipo b (bL y bH); el citocromo c tiene un grupo hemo tipo c (c1)), y la proteína hierro-azufre de Rieske posee una agrupación de dos átomos de hierro y dos de azufre (2Fe•2S).[2]
La ubiquinol-citocromo-c reductasa caltaliza la siguiente reacción química:
- QH2 + 2 ferricitocromo c Q + 2 ferrocitocromo c + 2 H+

Por lo tanto, los dos sustratos de esta enzima son la dehidroquinona (QH2) y el ferri- (Fe3+) citocromo c, mientras que sus tres productos son quinona, ferro- (Fe2+) citocromo c, y H+.
Pertenece, por lo tanto a la familia de las oxidorreductasas, específicamente a aquellas que actúan sobre los difenoles y sustancias relacionadas utilizándolas como dadores y al citocromo como aceptor.
Tiene una importante participación en la fosforilación oxidativa y cuenta con cuatro cofactores citocromo C1, citocromo b-562, citocromo b-566 y una ferredoxina de con dos átomos de hierro.
Nomenclatura
El nombre sistemático de esta clase de enzimas es ubiquinol:ferricitocromo-c oxidoreductasa. Otros nombres comúnmente utilizados son:
|
|
Estructura
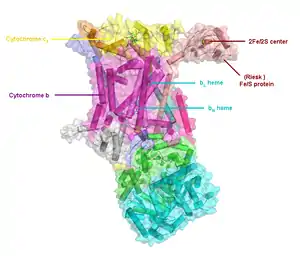
En comparación con las otras subunidades mayores bombeadoras de protones de la cadena de transporte de electrones, el número de subunidades que la componen puede parecer pequeño, tan pequeño como tres cadenas polipeptídicas, sin embargo este número puede aumentar y en los animales superiores se encuentran once subunidades.[4] Tres de estas subunidades poseen grupos prostéticos. La subunidad citocromo b posee dos grupos heme de tipo b (bL y bH), la subunidad citocromo c posee un grupo heme de tipo c, y la proteína de hierro azufre de Rieske (ISP) posee una agrupación formada por dos átomos de hierro y dos de azufre (2Fe•2S).
Reacción catalizada
El complejo III cataliza la reducción del citocromo c por medio de la reducción de la coenzima Q (CoQ), y el bombeo concomitante de 4 protones desde la matriz mitocondrial hacia el espacio intermembrana:
En el proceso conocido como ciclo Q,[5][6] se consumen dos protones de la matriz (M), se liberan cuatro protones hacia el espacio intermembrana (IM) y dos electrones pasan hacia el citocromo c.
Mecanismo de reacción
El mecanismo de reacción del complejo III (Citocromo bc1, Coenzima Q: Citocromo C oxidorreductasa) se conoce como el ciclo de la ubiquinona o ciclo Q. En este ciclo, cuatro protones son liberados en el lado positivo (P) de la membrana interna mitocondrial (hacia el espacio intermembrana), y se extraen cuatro protones desde la matriz (el lado negativo (N) de la membrana interna mitocondrial). En la reacción total, dos moléculas de ubiquinol se oxidan a ubiquinona, luego de lo cual una de las ubiquinonas se reduce a ubiquinol. Como resultado, dos electrones son transferidos desde el ubiquinol hacia la ubiquinona, por medio de dos citocromos c que actúan como intermediarios.
General:
- 2 x QH2 oxidados a Q
- 1 x Q reducidos a QH2
- 2 x Cyt c1 reducidos
- 4 x H+ son liberados al espacio intermembrana
- 4 x H+ son removidos de la matriz
La reacción procede de acuerdo a los siguientes pasos:
Primera ronda:
- El citocromo b une una molécula de ubiquinol y una de ubiquinona.
- El centro 2Fe/2S y el grupo heme bL quitan un electrón cada uno del ubiquinol unido, liberando dos hidrógenos en el espacio intermembrana.
- Uno de los electrones es transferido al citocromo c1 desde el centro 2Fe/2S, mientras que el otro electrón es transferido desde el grupo heme bL hacia el grupo heme bH.
- El citocromo c1 transfiere su electrón hacia el citocromo c (no debe ser confundido con el citocromo c1), y el heme bH transfiere su electrón hacia la cercana ubiquinona, resultando en la formación de una ubisemiquinona.
- El citocromo c difunde. La primera molécula de ubiquinol (que ahora se ha oxidado a ubiquinona) se libera, mientras que la semiquinona permanece unida.
Segunda ronda:
- Un segundo ubiquinol se une al citocromo b.
- El centro 2Fe/2S y el heme BL le quitan cada uno un electrón al ubiquinol recientemente unido, liberando dos hidrógenos hacia el espacio intermembrana.
- Un electrón es transferido desde el centro 2Fe/2S hacia el citocromo c1, mientras que otro electrón se transfiere desde el heme BL hacia el heme BH.
- El citocromo c1 luego transfiere su electrón hacia el citocromo c, mientras que la cercana semiquinona recoge un segundo electrón del heme BH, junto con dos protones de la matriz.
- El segundo ubiquinol (ahora oxidado a ubiquinona), se libera junto con el recientemente formado ubiquinol.[7]
Nombres de los genes humanos
MTCYB: mtDNA codifican para el citocromo b; las mutaciones de estos genes se encuentran relacionadas con la intolerancia al ejercicio
CYC1:citocromo c1
CYCS: citocromo c
UQCRFS1: proteína de Rieske
UQCRB: proteína de unión a la ubiquinona, las mutaciones de este gen se encuentran relacionadas con la deficiencia nuclear de complejo III mitocondrial tipo 3
UQCRH: proteína bisagra
UQCRC2: Núcleo 2, las mutaciones en este gen se encuentran relacionadas con la deficiencia de complejo III mitocondrial, de tipo 5 nuclear.
UQCRC1: Núcleo 1
UQCR: subunidad de 6.4KD
UQCR10: subunidad de 7.2KD
TTC19: Subunidad recientemente identificada, las mutaciones en este gen se encuentran relacionadas con la deficiencia de complejo III de tipo 2 nuclear.
Inhibidores del complejo III
Existen tres grupos distintivos de inhibidores del complejo III.
- La Antimicina A se une al sitio Qi e inhibe la transferencia de electrones en el complejo III desde el Heme bH al Q oxidado (inhibidor de sitio Qi).
- El Mixotiazol y la estigmatelina se unen al sitio Qo e inhiben la transferencia de electrones desde el QH2 reducido a la proteína de Rieske. El mixotiazol y la estigmatelina se unen a diferentes "bolsillos" en el sitio Qo.
- El mixotiazol se une a un punto muy cercano al citocromo bL (razón por la que se lo llama "inhibidor proximal").
- La estigmatelina se une a un sitio cercano a la proteína de Rieske, con la que interactúa fuertemente.
Algunos de los compuestos de estas familias han sido comercializados como funcigidas, (los derivados de la estrobilurina, de los cuales el mejor conocido es la azoxistrobina; inhibidor QoI) y como agentes antimaláricos (atovaquona).
Además la propilhexedrina también inhibe a la citocromo c reductasa.[8]
Especies reactivas de oxígeno
Una pequeña fracción de electrones abandonan la cadena de transporte electrónico antes de alcanzar al complejo IV. Una filtración prematura de electrones hacia el oxígeno resulta en la formación de superóxido. La relevancia de esta reacción (por lo demás minoritaria) es que el superóxido y otras especies reactivas de oxígeno son altamente tóxicas, y se cree que juegan un rol en diversas patologías, como así también en el envejecimiento, esto es lo que se conoce como teoría del envejecimiento por radicales libres.[9] La filtración de electrones ocurre principalmente en el sitio Qo y resulta estimulada por la antimicina A. La antimicina A bloquea a los hemes b en el estado reducido, previniendo su posterior reoxidación en el sitio Qi, lo que en cambio, provoca que las concentraciones basales de la semiquinona Qo se eleven, estas últimas especies reaccionan con el oxígeno para formar superóxido. Se cree que el alto potencial de membrana tiene un efecto similar.[10] El superóxido producido en el sitio Qo puede ser liberado tanto en la matriz mitocondrial,[11][12] como en el espacio intermembrana (desde donde después pueden alcanzar el citosol.[11][13] Esto puede ser explicado por el hecho de que el complejo III, podría producir superóxido en la forma de la especie permeable (capaz de atravesar la membrana hidrofóbica) hidroperoxilo (HOO•) en vez de la forma impermeable superóxido (O2-.).[12]
Mutaciones de genes del complejo III en enfermedades humanas
Las mutaciones en los genes emparentados con el Complejo III se manifiestan típicamente como diferentes tipos de intolerancia al ejercicio.[14][15] Se han reportado otras mutaciones que causan displasia septo-óptica[16] y enfermedades multisistémicas.[17] Sin embargo, las mutaciones en el gen BCS1L, uno de los genes responsables de la adecuada maduración del Complejo III, pueden resultar en un síndrome de Björnstad o GRACILE. Estos síndromes son letales en neonatos, en los cuales se desarrollan manifestaciones neurológicas y multisistémicas que tipifican varios síndromes mitocondriales severos. La patogenicidad de varias mutaciones han sido verificadas en sistemas biológicos modelo tales como las levaduras.[18]
Hasta el momento se desconoce cuál es la extensión de estas patologías debida a los defectos bioenergéticos o a la sobreproducción de superóxidos.
Véase también
Lecturas adicionales
- Marres CM, Slater EC (1977). «Polypeptide composition of purified QH2:cytochrome c oxidoreductase from beef-heart mitochondria». Biochim. Biophys. Acta. 462 (3): 531-548. PMID 597492. doi:10.1016/0005-2728(77)90099-8.
- Rieske JS (1976). «Composition, structure, and function of complex III of the respiratory chain». Biochim. Biophys. Acta. 456 (2): 195-247. PMID 788795.
- Wikstrom M, Krab K, Saraste M (1981). «Proton-translocating cytochrome complexes». Annu. Rev. Biochem. 50: 623-655. PMID 6267990. doi:10.1146/annurev.bi.50.070181.003203.
Referencias
- PDB 1ntz ; Gao X, Wen X, Esser L, Quinn B, Yu L, Yu CA, Xia D (agosto de 2003). «Structural basis for the quinone reduction in the bc1 complex: a comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors at the Qi site». Biochemistry 42 (30): 9067-80. PMID 12885240. doi:10.1021/bi0341814.
- Mathews, C. K.; Van Holde, K.E et Ahern, K.G (2003). Bioquímica (3 edición). ISBN 84-7892-053-2.
- Iwata S., Lee J.W., Okada K., Lee J.K., Iwata M., Rasmussen B., Link T.A., Ramaswamy S., Jap B.K. (1998) Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 281: 64-71
- Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK (julio de 1998). «Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex». Science 281 (5373): 64-71. PMID 9651245. doi:10.1126/science.281.5373.64.
- Kramer DM, Roberts AG, Muller F, Cape J, Bowman MK (2004). «Q-cycle bypass reactions at the Qo site of the cytochrome bc1 (and related) complexes». Meth. Enzymol. Methods in Enzymology 382: 21-45. ISBN 978-0-12-182786-1. PMID 15047094. doi:10.1016/S0076-6879(04)82002-0.
- Crofts AR (2004). «The cytochrome bc1 complex: function in the context of structure». Annu. Rev. Physiol. 66: 689-733. PMID 14977419. doi:10.1146/annurev.physiol.66.032102.150251.
- Ferguson SJ, Nicholls D, Ferguson S (2002). Bioenergetics (3rd edición). San Diego: Academic. pp. 114-117. ISBN 0-12-518121-3.
- http://www.ncbi.nlm.nih.gov/pubmed/241101
- Muller, F. L., Lustgarten, M. S., Jang, Y., Richardson, A. and Van Remmen, H. (2007). «Trends in oxidative aging theories». Free Radic. Biol. Med. 43 (4): 477-503. PMID 17640558. doi:10.1016/j.freeradbiomed.2007.03.034.
- Skulachev VP (mayo de 1996). «Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants». Q. Rev. Biophys. 29 (2): 169-202. PMID 8870073.
- Muller F (2000). «The nature and mechanism of superoxide production by the electron transport chain: Its relevance to aging». AGE 23 (4): 227-253. doi:10.1007/s11357-000-0022-9.
- Muller FL, Liu Y, Van Remmen H (noviembre de 2004). «Complex III releases superoxide to both sides of the inner mitochondrial membrane». J. Biol. Chem. 279 (47): 49064-73. PMID 15317809. doi:10.1074/jbc.M407715200.
- Han D, Williams E, Cadenas E (enero de 2001). «Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space». Biochem. J. 353 (Pt 2): 411-6. PMC 1221585. PMID 11139407. doi:10.1042/0264-6021:3530411.
- DiMauro S (noviembre de 2006). «Mitochondrial myopathies». Curr Opin Rheumatol 18 (6): 636-41. PMID 17053512. doi:10.1097/01.bor.0000245729.17759.f2.
- DiMauro S (junio de 2007). «Mitochondrial DNA medicine». Biosci. Rep. 27 (1–3): 5-9. PMID 17484047. doi:10.1007/s10540-007-9032-5.
- Schuelke M, Krude H, Finckh B, Mayatepek E, Janssen A, Schmelz M, Trefz F, Trijbels F, Smeitink J (marzo de 2002). «Septo-optic dysplasia associated with a new mitochondrial cytochrome b mutation». Ann. Neurol. 51 (3): 388-92. PMID 11891837. doi:10.1002/ana.10151.
- Wibrand F, Ravn K, Schwartz M, Rosenberg T, Horn N, Vissing J (octubre de 2001). «Multisystem disorder associated with a missense mutation in the mitochondrial cytochrome b gene». Ann. Neurol. 50 (4): 540-3. PMID 11601507. doi:10.1002/ana.1224.
- Fisher N, Castleden CK, Bourges I, Brasseur G, Dujardin G, Meunier B (marzo de 2004). «Human disease-related mutations in cytochrome b studied in yeast». J. Biol. Chem. 279 (13): 12951-8. PMID 14718526. doi:10.1074/jbc.M313866200.
Enlaces externos
- MeSH: Coenzyme+Q-Cytochrome-c+Reductase (en inglés)
- sito del complejo citocromo bc1 (Edward A. Berry) en lbl.gov (inglés)
- cytochrome sitio del complejo bc1 (Antony R. Crofts) en uiuc.edu
- PROMISE Database: cytochrome bc1 complex at scripps.edu
- Modelo molecular interactivo del Complejo III (Requiere MDL Chime)
| Control de autoridades |
|
|---|
 Datos: Q412832
Datos: Q412832 Multimedia: Coenzyme Q – cytochrome c reductase / Q412832
Multimedia: Coenzyme Q – cytochrome c reductase / Q412832