Hemo
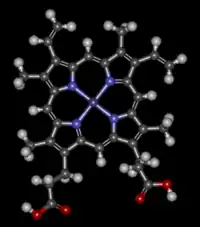
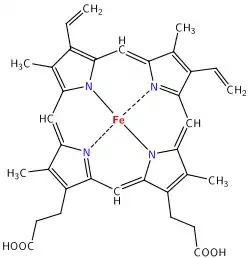
El grupo hemo (del griego αἷμα "sangre") es un grupo prostético que forma parte de diversas proteínas, entre las que destaca la hemoglobina, consiste en un ion Fe2+
(ferroso) contenido en el centro de un gran heterociclo orgánico llamado porfirina, hecho de cuatro grupos pirrólicos unidos entre sí por medio de puentes metileno. No todas las porfirinas contienen hierro, pero una fracción sustancial de las metaloproteínas que contienen el núcleo porfirina, poseen el grupo hemo como grupo prostético; estas proteínas se conocen como hemoproteínas. El grupo hemo es principalmente conocido por formar parte de la hemoglobina, el pigmento rojo de la sangre, pero también se encuentra en un gran número de otras hemoproteínas biológicamente importantes tales como la mioglobina, citocromos, catalasa, y la óxido nítrico sintasa endotelial.

) en el centro de la protoporfirina IX.
Función

Las hemoproteínas poseen diversas funciones biológicas, incluyendo el transporte de gases diatómicos, catálisis química, y detección de gases diatómicos y transferencia de electrones. El ion hemo sirve como fuente o sumidero de electrones durante transferencias electrónicas o reacciones redox. En las reacciones de las peroxidasas, la molécula de porfirina sirve además como fuente de electrones. En el transporte o detección de gases diatómicos, el gas se une al ion hemo. Durante la detección de gases diatómicos, la unión del gas ligando al grupo hemo induce cambios conformacionales en la proteína que lo rodea.
Se ha especulado que la función evolutiva original de las hemoproteínas fue la transferencia de electrones en la fotosíntesis primitiva basada en los compuestos de azufre que realizaban los organismos similares a cianobacterias ancestrales, antes de que apareciera el oxígeno molecular.[1]
Las hemoproteínas han alcanzado su remarcable diversidad funcional modificando el ambiente inmediato del macrociclo hemo dentro de la matriz proteica. Por ejemplo, la capacidad de la hemoglobina para entregar en forma efectiva el oxígeno a los tejidos se debe a unos residuos aminoacídicos específicos localizados cerca del grupo hemo de la molécula. La hemoglobina une oxígeno en la vasculatura pulmonar, donde el pH es alto y la pCO
2 es baja, y lo libera en los tejidos, donde la situación se invierte. Este fenómeno se conoce como efecto Bohr. El mecanismo molecular detrás de este efecto es la organización estérica de la cadena globina; un residuo de histidina localizado en una posición adyacente al grupo hemo, deviene en positivamente cargado cuando el pH se acidifica (lo cual es causado por la disolución del dióxido de carbono en tejidos con alta tasa metabólica), liberando estéricamente al oxígeno del grupo hemo.
Estructura química
El grupo hemo contiene hierro y un anillo de porfirina; el que corresponde a un tetrapirrol cíclico, este macrociclo está compuesto por 4 anillos de pirrol unidos por puentes metino (=CH-) o a veces mal llamado metileno (=CH
2), en el centro de este anillo se encuentra el átomo de hierro (II) tetracoordinado por los cuatro pares de electrones no compartidos de los nitrógenos del anillo porfirina.
Tipos
Principales hemos
Hay varios tipos de grupos hemo biológicamente importantes:
| Hemo A | Hemo B | Hemo C | Hemo O | ||
|---|---|---|---|---|---|
| Número PubChem | 7888115 | 444098 | 444125 | 6323367 | |
| Fórmula química | C 49H 56O 6N 4Fe |
C 34H 32O 4N 4Fe |
C 34H 36O 4N 4S 2Fe |
C 49H 58O 5N 4Fe | |
| Grupo funcional en C 3 |
 |
-CH(OH)-CH 2-Far |
-CH=CH 2 |
-CH(cisteín-S-il)-CH 3 |
-CH(OH)-CH 2-Far |
| Grupo funcional en C 8 |
-CH=CH 2 |
-CH=CH 2 |
-CH(cisteín-S-il)-CH 3 |
-CH=CH 2 | |
| Grupo funcional en C 18 |
-CH=O | -CH 3 |
-CH 3 |
-CH 3 | |


El tipo de Hemo más común en la naturaleza es el Hemo B; otros tipos importantes son el Hemo A y el Hemo C. Los grupos heme aislados comúnmente se designan con letras mayúscula, mientras que los grupos heme unidos a las proteínas se designan con las letras en minúscula. El citocromo a se refiere al grupo hemo A en una combinación específica con una proteína de membrana para formar una porción de la citocromo c oxidasa.
Otros hemos
- Nota:El sistema de numeración de carbonos utilizado a continuación es el viejo sistema utilizado por los bioquímicos, no el sistema 1-24 recomendado por la IUPAC que se muestra en la tabla más arriba.
- Hemo l es un derivado del Hemo B que se encuentra covalentemente unido a la proteína en la lactoperoxidasa, peroxidasa de eosinófilo y peroxidasa de tiroides. La adición de peróxido con los residuos glutamil-375 y aspartil-225 de la lactoperoxidasa forma enlaces tipo ester entre estos aminoácidos y los grupos metil 1 y 5 respectivamente del grupo hemo.[4][5]
Se piensa que se forman enlaces éster similares con estos dos grupos metil en las peroxidasas de eosinófilo y de tiroides. El grupo Hemo l es una característica importante de las peroxidasas animales; las peroxidasas de plantas incorporan Hemo B. La lactoperoxidasa y la peroxidasa de eosinófilo son enzimas protectoras, que forman parte del mecanismo de defensa del organismo contra otros organismos invasores. La peroxidasa de tiroides es la enzima que cataliza la biosíntesis de las importantes hormonas tiroideas. Debido a que la lactoperoxidasa destruye organismos invasores en los pulmones y en los excrementos, se piensa que desempeña un importante papel protector.
- Hemo m es un derivado del Hemo B unido covalentemente al sitio activo de la mieloperoxidasa. El Hemo m contiene dos enlaces tipo éster en las posiciones 1 y 5 metilo, como ocurre en el Hemo l encontrado en otras peroxidasas de mamíferos. Además, posee un único ion sulfonio unido entre el azufre de un residuo aminoácido y el grupo vinilo en C2, dándole a esta enzima la capacidad de oxidar con facilidad iones cloruro y bromuro, para formar hipoclorito e hipobromito dos compuestos con una altísima capacidad microbicida. La mieloperoxidasa se encuentra presente en los neutrófilos animales y es responsable por la destrucción de bacterias y virus. Sintetiza hipobromito casi "por error", ya que es un compuesto mutagénico, pero la cantidad de iones bromuro presentes en los tejidos es muy baja.
- Hemo D es otro derivado del Hemo B, pero en el cual el ácido propiónico de la cadena lateral en C6, el cual se encuentra además hidroxilado, forma un anillo γ-espirolactona. El anillo III se encuentra además hidroxilado en la posición 5, en una conformación trans al nuevo grupo lactona.[5] El Hemo D es el sitio donde se produce la reducción del oxígeno a agua en muchos tipos de bacterias que funcionan a una baja tensión de oxígeno.
- Hemo S está relacionado con el Hemo B por la presencia de un grupo formilo en la posición 2 en lugar del grupo 2-vinilo. El Hemo S se encuentran en la hemoglobina de los gusanos marinos. La correcta estructura de los Hemo B y S fue dilucidada por primera vez por el químico alemán Hans Fischer.
Los nombres de los citocromos típicamente (aunque no siempre) reflejan el tipo de grupo heme qu contienen así por ejemplo el citocromo a contiene Hemo A, el citocromo c contiene Hemo C, etc.
Síntesis
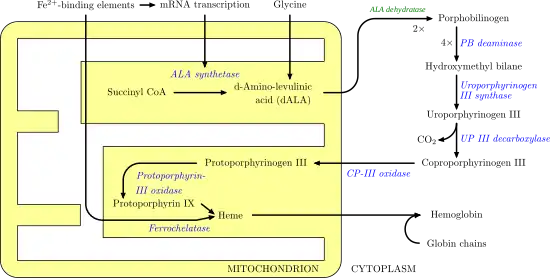
El proceso enzimático que lleva a la producción del grupo hemo, se llama apropiadamente porfirinosíntesis, ya que todos los intermediarios son tetrapirroles se clasifican químicamente como porfirinas.
El proceso se encuentra altamente conservado en todos los seres vivos. En humanos, esta vía metabólica sirve casi exclusivamente para la síntesis del hemo. En otras especies, además produce sustancias similares tales como la cobalamina.
La vía se inicia con la síntesis de ácido δ-aminolevulínico (dALA o δALA) a partir del aminoácido glicina y de succinil-CoA proveniente del ciclo del ácido cítrico. La enzima limitante de velocidad en esta reacción, la ALA sintasa, se encuentra regulada negativamente por la concentración de glucosa y hemo. Los mecanismos de inhibición de la ALAs por hemo o hemina se produce por medio de la disminución de la estabilidad de la síntesis de ARNm y por la disminución de la incorporación de ARNm en la mitocondria. Este mecanismo tiene una importancia terapéutica, la infusión de arginato de heme o hematina y glucosa puede abortar los ataques de porfiria intermitente aguda en pacientes con un error innato del metabolismo en este proceso. Y funciona reduciendo la transcripción de la ALA sintasa.[6]
Aunque todas las células precisan del grupo hemo para funcionar adecuadamente, los órganos principalmente involucrados en la síntesis del hemo son el hígado (en el cual la síntesis de hemo es altamente variable, dependiendo del contenido global de hemo del organismo) y la médula ósea (en la cual la tasa de producción de heme es relativamente constante, y depende de la producción de la cadena de globina). El hemo puede ser visto como una molécula intermediaria en el catabolismo de la hemoglobina que conduce a la producción de bilirrubina. Defectos en varias enzimas que participan en la síntesis del hemo pueden conducir a un grupo de enfermedades llamadas porfirias, entre las que se incluyen: porfiria intermitente aguda, porfiria congénita eritropoyética, porfiria cutánea tarda, coproporfiria hereditaria, porfiria variegata y la protoporfiria eritropoyética.
Degradación

La degradación del grupo hemo comienza dentro de los macrófagos del bazo, los cuales retiran los eritrocitos senescentes de la circulación. En un primer paso, el grupo hemo se convierte en biliverdina, por la enzima hemo oxigenasa (HMOX). Se utiliza NADPH como agente reductor, se introduce oxígeno a la reacción y se libera monóxido de carbono (CO), mientras que el hierro que se libera de la molécula lo hace en la forma de ion férrico (3+). El monóxido de carbono actúa como mensajero celular y tiene alguna función en la vasodilatación.
Adicionalmente, la degradación del heme parece ser una respuesta evolutivamente muy conservada al estrés oxidativo. Brevemente, cuando una célula es expuesta a radicales libres, se produce una rápida inducción de la expresión de la HMOX1 la cual es una hemo oxigenasa de respuesta al estrés. Esta isoenzima cataboliza a los grupos hemo. La razón por la cual las células podrían aumentar exponencialmente su capacidad de degradar el grupo hemo en respuesta al estrés oxidativo todavía permanece poco clara, pero parece formar parte de una respuesta citoprotectora que limita los efectos deletéreos del hemo libre.
HMOX1/2
hemo -------------- biliverdina + Fe3+
/ \
H+
+ NADPH NADP+
O
2 CO
En la segunda reacción, la biliverdina se convierte en bilirrubina, por acción de la biliverdina reductasa (BVR):
BVR
biliverdina ----------- bilirrubina
/ \
H+
+ NADPH NADP+
La bilirrubina se transporta al hígado unida a proteína (albúmina sérica), donde se conjuga con ácido glucurónico para hacerla más soluble en agua. La reacción es catalizada por la enzima UDP-glucurónido transferasa (UDPGUTF).
UDPGUTF
bilirrubina + 2 UDP-glucuronato ------------ diglucurónido de bilirrubina
\
2 UMP + 2 Pi
Esta forma de bilirrubina se excreta del hígado a través de la bilis. La excesión de bilirrubina hacia los canalículos biliares es un proceso activo, dependiente de energía y limitante. La flora intestinal desconjuga al diglucurónido de bilirrubina, y convierte a la bilirrubina en urobilinógenos. Una parte de este urobilinógeno se reabsorbe en el intestino y viaja por sangre hasta que es excretado con la orina por los riñones en forma de urobilina, la cual es un producto de la oxidación del urobilinógeno, y es la que le da el color amarillo a la orina. El urobilinógeno restante viaja por el tracto digestivo donde se convierte en estercobilinógeno. Este se oxida a estercobilina, la cual es la responsable del color oscuro de las heces.
El grupo hemo en la salud y en la enfermedad
Bajo condiciones de homeostasis, la reactividad del grupo hemo se encuentra bajo control, ya que se encuentra insertado dentro de los "bolsillos hemo" de las hemoproteínas. Sin embargo, bajo condiciones de estrés oxidativo, algunas hemoproteínas, tales como por ejemplo la hemoglobina, pueden liberar sus grupos prostéticos.[7][8] El hemo no proteico (libre) que se produce de esta manera es altamente tóxico, más probablemente debido al átomo de hierro contenido dentro del anillo protoporfirina IX, el cual actúa como un reactivo de Fenton para catalizar la producción de radicales libres.[cita requerida] Esta propiedad del hemo libre puede sensibilizar a una variedad de células para entrar en muerte celular programada en respuesta a agonistas proinflamatorios, un efecto deletéreo que juega un importante rol en la patogénesis de ciertas enfermedades inflamatorias tales como la malaria[9] y sepsis.[10]
Genes
Los siguientes genes son parte de la vía metabólica de síntesis del heme:
- ALAD: ácido aminolevulínico, delta-, deshidratasa (la deficiencia causa porfiria por deficiencia de ALA-deshidratasa)[11]
- ALAS1: aminolevulinato, delta-, sintasa 1
- ALAS2: aminolevulinato, delta-, sintasa 2 ((la deficiencia causa anemia sideroblástica/hipocrómica)
- CPOX: coproporfirinógeno oxidasa ((la deficiencia causa coproporfiria hereditaria)[12]
- FECH: protoporfiria por deficiencia de ferroquelatasa
- HMBS: hidroximetilbilano sintasa ((la deficiencia causa porfiria intermitente aguda)[13]
- PPOX: protoporfirinógeno oxidasa ((la deficiencia provoca porfiria variegata)[14]
- UROD: uroporfirinógeno descarboxilasa ((la deficiencia provoca porfiria cutánea tardía) [15]
- UROS: uroporfirinógeno III sintasa (la deficiencia provoca porfiria eritropoyética congénita)
Véase también
Referencias
- Hardison, R. (1999). «The Evolution of Hemoglobin: Studies of a very ancient protein suggest that changes in gene regulation are an important part of the evolutionary story». American Scientist 87 (2): 126. Archivado desde el original el 10 de abril de 2016.
- Caughey, W. S.; Smythe, G. A.; O'Keeffe, D. H.; Maskasky, J. E.; Smith, M. I. (1975). «Heme A of cytochrome c oxicase. Structure and properties: comparisons with hemes B, C, and S and derivatives». J. Biol. Chem. 250 (19): 7602-7622. PMID 170266.
- Brown, K. R.; Brown, B. M.; Hoagland, E.; Mayne, C. L.; Hegg, E. L. (2004). «Heme A Synthase Does Not Incorporate Molecular Oxygen into the Formyl Group of Heme A». Biochemistry 43 (27): 8616-8624. PMID 15236569. doi:10.1021/bi049056m.
- Rae, T. D.; Goff, H. M. (1998). «The heme prosthetic group of lactoperoxidase. Structural characteristics of heme l and heme l-peptides». J. Biol. Chem. 273 (43): 27968-77. PMID 9774411.
- Murshudov, G. N.; Grebenko, A. I.; Barynin, V.; Dauter, Z.; Wilson, K. S.; Vainshtein, B. K.; Melik-Adamyan, W; Bravo, J. et al. (1996). «Structure of the heme d of Penicillium vitale and Escherichia coli catalases». J. Biol. Chem. 271 (15): 8863-8. PMID 8621527.
- Kolluri, Sridevi (17 de marzo de 2004). Upregulation of Heme Pathway Enzyme ALA Synthase-1 by Glutethimide and 4,6-Dioxoheptanoic Acid and Downregulation by Glucose and Heme: A Dissertation (en inglés estadounidense). doi:10.13028/yyrz-qa79. Consultado el 26 de febrero de 2023.
- Bunn, H. F.; Jandl, J. H. (1966). «Exchange of heme among hemoglobin molecules.». Proc Natl Acad Sci U S A 56 (3): 974-8. PMID 5230192.
- Smith, M. L.; Paul, J.; Ohlsson, P. I.; Hjortsberg, K.; Paul, K. G. (1991). «Heme-protein fission under nondenaturing conditions». Proc Natl Acad Sci U S A 88 (3): 882-6. PMID 1846966.
- Pamplona, A.; Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Epiphanio, S.; Chora, A.; Rodrigues, C. D. et al. (June 2007). «Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria.». Nature Medicine 13 (6): 703-710. PMID 17496899. doi:10.1038/nm1586.
- Larsen, Rasmus; Gozzelino, Raffaella; Jeney, Viktória; Tokaji, László; Bozza, Fernando A.; Japiassú, André M.; Bonaparte, Dolores; Cavalcante, Moisés Marinho; Chora, Angelo; Ferreira, Ana; Marguti, Ivo; Cardoso, Sílvia; Sepúlveda, Nuno; Smith, Ann; Soares, Miguel P. (29 de septiembre de 2010). «A central role for free heme in the pathogenesis of severe sepsis». Science Translational Medicine 2 (51): 51ra71. ISSN 1946-6242. PMID 20881280. doi:10.1126/scitranslmed.3001118. Consultado el 8 de agosto de 2016.
- Plewinska, Magdalena; Thunell, Stig; Holmberg, Lars; Wetmur, James; Desnick, Robert (1991). «delta-Aminolevulinate dehydratase deficient porphyria: identification of the molecular lesions in a severely affected homozygote». American Journal of Human Genetics (49): 167-174. PMC 1683193. PMID 2063868. Consultado el 18 de noviembre de 2014.
- Aurizi, C; Lupia Palmieri, G; Barbieri, L; Macri, A; Sorge, F; Usai, G; Biolcati, G (February 2009). «Four novel mutations of the coproporphyrinogen III oxidase gene». Cellular and Molecular Biology 55 (1): 8-15. PMID 19267996. Consultado el 18 de noviembre de 2014.
- Bustad, HJ; Vorland, M; Ronneseth, E; Sandberg, S; Martinez, A; Toska, K (8 de agosto de 2013). «Conformational stability and activity analysis of two hydroxymethylbilane synthase mutants, K132N and V215E, with different phenotypic association with acute intermittent porphyria». Bioscience Reports 33 (4). doi:10.1042/BSR20130045. Consultado el 18 de noviembre de 2014.
- Martinez di Montemuros, F; Di Pierro, E; Patti, E; Tavazzi, D; Danielli, MG; Biolcati, G; Rocchi, E; Cappllini, MD (Diciembre de 2002). «Molecular characterization of porphyrias in Italy: a diagnostic flow-chart». Cellular and Molecular Biology 48 (8): 867-876. PMID 12699245. Consultado el 18 de noviembre de 2014.
- Badenas, C; To-Figueras, J; Phillips, JD; Warby, CA; Munoz, C; Herrero, C (Abril de 2009). «Identification and characterization of novel uroporphyrinogen decarboxylase gene mutations in a large series of porphyria cutanea tarda patients and relatives». Clinical Genetics 75 (4): 346-353. PMC 3804340. PMID 19419417. doi:10.1111/j.1399-0004.2009.01153.x. Consultado el 18 de noviembre de 2014.
Enlaces externos
 Wikimedia Commons alberga una categoría multimedia sobre el grupo hemo.
Wikimedia Commons alberga una categoría multimedia sobre el grupo hemo.
 Datos: Q189621
Datos: Q189621