Cicatrización
La cicatrización es un proceso biológico mediante el cual los tejidos vivos reparan sus heridas dejando para el caso de las heridas cutáneas, una cicatriz que puede ser estética o inestética.[1]
Cuando una persona sufre una herida, en el proceso de reparación se llevan a cabo una serie de complejas reacciones bioquímicas que suceden para reparar el daño. Estos fenómenos ocurren con cierto solapamiento temporal y pueden ser divididos para su estudio en las siguientes fases: inflamatoria, proliferativa, y de remodelación (algunos autores consideran que la cicatrización ocurre en cuatro o más etapas, si se subdividen las fases inflamatoria o de proliferación en pasos intermedios).[2][3][4][3]
| Cicatrización de una herida en una mano | ||||
 |
 |
 |
 | |
| Días después de producida la herida | ||||
| 0 | 2 | 17 | 30 | |
Fases de la cicatrización
En la fase inflamatoria, se fagocitan y eliminan las bacterias, la suciedad, y se liberan factores que producen la migración y división de las células que toman parte en la fase proliferativa.
La fase proliferativa se caracteriza por la angiogénesis, el aumento de colágeno, la formación de tejido granular, la epitelización, y la contracción de la herida.[5] En la angiogénesis, crecen nuevos vasos sanguíneos a partir de células endoteliales.[6] En la fibroplasia y formación de tejido granular, los fibroblastos crecen y forman una nueva matriz extracelular provisoria (ECM, por las siglas en inglés: ExtraCellular Matrix) mediante la secreción de colágeno y fibronectina.[5] En la epitelialización, las células epiteliales se desplazan sobre la herida cubriéndola.[7] En la contracción, los miofibroblastos ayudan a reducir el tamaño la herida; ellos se adhieren de los bordes de la herida y se contraen utilizando un mecanismo similar al que tienen las células de los músculos lisos. Cuando las células han cumplido con su cometido, las células no utilizadas sufren una apoptosis.[5]
En la fase de maduración y remodelado, el colágeno es remodelado y realineado a lo largo de las líneas de tensión y las células que ya no se precisan son eliminadas mediante una apoptosis.
Sin embargo, este proceso no solo es complejo sino que es frágil y es susceptible de ser interrumpido o fallar, lo que conduce a la formación de heridas crónicas con problemas de cicatrización. Algunos factores que pueden contribuir a este problema son la diabetes, enfermedades de las venas o arterias, edad avanzada, e infecciones.[8]
Fase inflamatoria
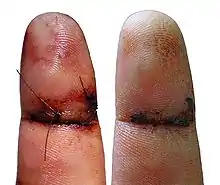
Durante la fase inflamatoria, ocurre un proceso de coagulación que detiene la pérdida de sangre (hemostasia), además se liberan varios factores para atraer células que fagociten; residuos, bacterias, tejido dañado y liberen factores que inicien la fase proliferativa de cicatrización de la herida.
Cascada de coagulación
Cuando un tejido biológico es herido, la sangre toma contacto con el colágeno, lo que provoca que las plaquetas de la sangre comiencen a secretar factores inflamatorios.[9] Las plaquetas también producen glicoproteínas en sus membranas celulares que les permiten adherirse unas a otras, formando una masa.[5]
La fibrina y la fibronectina se enlazan y forman una red o tapón que atrapa proteínas y partículas evitando de esta manera que continúe la pérdida de sangre[10] Este tapón de fibrina-fibronectina se constituye también en el principal soporte estructural de la herida hasta tanto se deposite el colágeno.[5] Las células migratorias utilizan este tapón como una matriz que les ayuda a desplazarse, las plaquetas se adhieren a la misma y secretan diversos factores.[5] El coágulo es eventualmente degradado por lisinas y reemplazado por tejido granular y posteriormente por colágeno.
Plaquetas
Las plaquetas son fragmentos de células (megacariocitos) que intervienen en el proceso de coagulación, confluyen en mayor número al producirse una herida y liberan una serie de sustancias en la sangre, incluidas proteínas ECM, citoquinas, y factores de crecimiento.[9] Los factores de crecimiento estimulan a las células para que aumenten su velocidad de división. Las plaquetas también liberan otros factores que favorecen la inflamación tales como son la serotonina, bradiquinina, prostaglandinas, prostaciclinas, tromboxano, e histamina; que aumentan la velocidad y migración de células hacia la zona, favorecen a los vasos sanguíneos en el proceso de dilatación y aumento de porosidad.[2]
Vasoconstricción y vasodilatación
Inmediatamente después de que resulte dañado un vaso sanguíneo, las membranas celulares dañadas liberan factores inflamatorios tales como tromboxanos y prostaglandinas, éstos hacen que el vaso se contraiga minimizando la pérdida de sangre y ayudando a que se aglutinen en el área las células inflamatorias y los factores inflamatorios.[2] Esta vasoconstricción dura de cinco a diez minutos y es seguida por una etapa de vasodilatación, en la cual se expanden los vasos sanguíneos, fenómeno que alcanza su máximo unos veinte minutos después de haberse producido la herida.[2] La vasodilatación es producida por varios factores liberados por las plaquetas y otras células. El principal factor que desencadena la vasodilatación es la histamina.[2][9] La histamina también hace que los vasos sanguíneos se tornen porosos, lo que permite que el tejido se vuelva edematoso a causa de las proteínas que aporta el torrente sanguíneo al espacio extravascular, lo cual aumenta la carga osmolar y aporta agua a la zona.[2] El incremento de la porosidad en los vasos sanguíneos también facilita la entrada de células inflamatorias, tales como leucocitos, en la zona de la herida desde el torrente sanguíneo.[11][12]
Leucocitos polimorfonucleares
Al cabo de una hora de haberse producido la herida, los leucocitos polimorfonucleares o granulocitos llegan a esta y se convierten en las células más abundantes en la zona de la herida durante los próximos tres días. Es particularmente elevada su cantidad durante el segundo día.[13] La fibronectina, los factores de crecimiento, y substancias tales como neuropéptidos y quininas son los que los atraen a la herida. Los granulocitos fagocitan los residuos y bacterias, aunque también matan a las bacterias mediante la liberación de radicales libres en un proceso denominado 'respiratory burst'.[14][15] También limpian la herida mediante la secreción de proteasas que rompen el tejido dañado. Una vez que han completado su tarea los granulocitos sufren un proceso de apóptosis y son devorados, degradados por los macrófagos.[16]
Otros leucocitos que se encuentran en la zona son células T ayudantes, que secretan citoquinas para inducir la subdivisión de las células T, aumentar la inflamación, mejorar la vasodilatación y permeabilidad de los vasos.[11][17] Las células T también aumentan la actividad de los macrófagos.[11]
Macrófagos
Los macrófagos son células que tienen función fagocitaria, por lo tanto son esenciales para la cicatrización de una herida.[13] Después de transcurridos dos días de producida la herida, los macrófagos son las células más abundantes en la zona de la herida.[18] Los monocitos del torrente sanguíneo son atraídos a la zona de la herida por los factores de crecimiento liberados por las plaquetas y otras células, los monocitos penetran la zona de la herida atravesando las paredes de los vasos sanguíneos.[19] La presencia de monocitos en la herida alcanza su máxima proporción después de 24 a 36 horas de haberse producido la herida.[17] Una vez que se encuentran en la zona de la herida, los monocitos maduran y se transforman en macrófagos, que es la principal célula responsable de limpiar la zona de bacterias y residuos.[13]
El principal rol de los macrófagos es fagocitar bacterias y al tejido dañado, también el último mediante la liberación de proteasas.[20] Los macrófagos secretan ciertos factores tales como factores de crecimientos y otras citoquinas, especialmente unos tres a cuatro días luego de producida la herida. Dichos factores atraen al área a células que participan en la etapa de proliferación de cicatrización de la herida.[9] El bajo contenido de oxígeno en la zona estimula a los macrófagos, a producir factores que inducen e incrementan la velocidad de angiogénesis.[14] y también estimulan a las células a producir la reepitelización de la herida, crear tejido granular, y formar una nueva matriz extracelular.[21][22] La capacidad de los macrófagos para secretar estos factores, los convierte en elementos vitales para promover que el proceso de cicatrización de la herida evolucione a la fase siguiente.
La inflamación es una parte necesaria del proceso de cicatrización, dado que cumple ciertos roles en el combate de la infección e inducción de la fase de proliferación. Sin embargo, si la inflamación se prolonga durante mucho tiempo puede producir daño a los tejidos.[5] Por esta razón, la reducción de la inflamación es frecuentemente un objetivo de los cuidados terapéuticos. La inflamación continúa mientras existan residuos en la herida. Por ello la presencia de residuos u otros objetos puede extender más allá de lo conveniente la fase de inflamación, dando eventualmente origen a una herida crónica.
Al ir desapareciendo la inflamación, se reduce la secreción de factores de inflamación, los factores existentes son eliminados y disminuye la presencia de neutrófilos y macrófagos en la zona de la herida.[13] Estos cambios dan indicio de la finalización de la fase de inflamación y el comienzo de la fase proliferativa.[13]
Fase proliferativa
Después de transcurridos dos a tres días desde la ocurrencia de la herida, comienza la afluencia de fibroblastos en la cicatriz, marcando el comienzo de la fase proliferativa aún antes que la fase inflamatoria haya concluido.[23] Al igual que las otras fases de la cicatrización, los pasos en la fase proliferativa no tienen lugar de forma sucesiva sino que los mismos ocurren simultáneamente.
Angiogénesis
El proceso de angiogénesis (también llamado neovascularización) tiene lugar simultáneamente con la proliferación de fibroblastos, cuando las células endoteliales migran hacia la zona de la herida.[24] La angiogénesis es imprescindible para otras etapas del proceso de cicatrización, tales como la migración epidérmica y de fibroblastos, aportando el oxígeno que precisan los últimos y células epiteliales para desarrollar sus funciones. El tejido en que se desarrolla la angiogénesis posee un color rojo (es eritematoso) producto de la presencia de capilares sanguíneos.[24]
Para poder generar nuevos vasos sanguíneos y alimentar con oxígeno y nutrientes al tejido las células madres llamadas células endoteliales provenientes de vasos sanguíneos no dañados desarrollan pseudópodos y se desplazan a través del ECM hacia la zona de la herida.[25] Al realizar esta actividad, ellas generan nuevos vasos sanguíneos.[14]
Para migrar, las células endoteliales necesitan colagenasas y activadores plasminogénicos para disolver el coágulo y parte del ECM[2][13] metaloproteinasas basadas en zinc digieren la membrana basal y el ECM para permitir la proliferación de células y la angiogénesis.[26]
Las células endoteliales también son atraídas hacia la zona de la herida por la fibronectina que se encuentra en el scab de fibrina y por factores de crecimiento secretados por otras células.[25] El crecimiento endotelial y la proliferación son también estimulados por la hipoxia y presencia de ácido láctico en la herida.[23] En un medio ambiente con bajo contenido de oxígeno, los macrófagos y plaquetas producen factores angiogénicos que atraen la células endoteliales mediante quimiotaxis. Cuando el medio en que se encuentran los macrófagos y otros células productoras de factores de crecimiento deja de ser hipóxico y de estar saturado de ácido láctico, dejan de producir factores angiogénicos.[14] Por lo tanto, cuando el tejido es perfundido en forma adecuada, se reduce la migración y proliferación de células endoteliales. Eventualmente aquellos vasos sanguíneos que ya no se precisan mueren mediante apoptosis.[25]
Fibroplasia y formación de tejido granular
De forma simultánea con la angiogénesis comienza la acumulación de fibroblastos en la zona de la herida. Los fibroblastos comienzan a aparecer dos a cinco días después de producida la herida. Cuando la fase inflamatoria está finalizando su número alcanza un máximo entre una a dos semanas después de producida la herida.[13] Hacia el final de la primera semana los fibroblastos son las células que se presentan con mayor abundancia en la citatriz.[2] La fibroplasia finaliza después de unas dos a cuatro semanas de ocurrida esta.
Durante los primeros dos a tres días después de producida la herida, los fibroblastos proliferan y migran, mientras que posteriormente ellos son las principales células responsables de generar la matriz de colágeno en la cicatriz.[2] Los fibroblastos que se encuentran en el tejido normal migran hacia la zona de la herida desde sus márgenes. Inicialmente los fibroblastos utilizan el fibrina scab formado en la fase inflamatoria para migrar, adhiriéndose a la fibronectina.[25] Los fibroblastos depositan inicialmente sustancia basal en la base de la herida y posteriormente colágeno, al cual se pueden adherir para migrar.[9]
El tejido granular es necesario para rellenar el agujero que ha dejado una herida que atraviesa la membrana basal. Comienza a hacer su aparición en la cicatriz durante la fase inflamatoria, unos dos a cinco días después de ocurrida la herida, y continúa creciendo hasta que se cubre la base de esta. El tejido granular se compone de nuevos vasos sanguíneos; fibroblastos, células inflamatorias, células endoteliales, miofibroblastos y los componentes de un nuevo ECM provisorio. La composición del ECM provisorio es diferente de la composición del ECM en el tejido normal e incluye fibronectina, colágeno, glicosaminoglicanos, y proteoglicanos.[25] Sus principales componentes son fibronectina y hialuronano, los cuales crean una matriz altamente hidratada que facilita la migración de las células.[19] Posteriormente esta matriz provisoria es reemplazada por un ECM que posee mayores similitudes que aquella que se encuentra en tejidos sin heridas.
Los fibroblastos depositan moléculas ECM como ser glicoproteínas, glicosaminoglicanos (GAGs), proteoglicanos, elastina, y fibronectina, que después utilizan para migrar a través de la herida (Cohen, 2005).
Los factores de crecimiento (PDGF, TGF-β) y la fibronectina promueven la proliferación y la migración hacia la base de la herida y la producción de moléculas ECM por los fibroblastos. Los fibroblastos también secretan factores de crecimiento que atraen células epiteliales hacia la cicatriz. La hipoxia también contribuye a la proliferación de los fibroblastos y la producción de factores de crecimiento, si bien una baja concentración de oxígeno inhibirá su crecimiento y la deposición de componentes ECM y puede producir una cicatriz excesivamente fibrosa.
Disposición de colágeno
Una de las tareas más importantes de los fibroblastos es la producción de colágeno.[24] Los fibroblastos comienzan a secretar una cantidad importante de este dos a tres días después de producida la herida, y su disposición alcanza su máximo de una a tres semanas después.[25][21] La producción de colágeno continúa a buen ritmo durante dos a cuatro semanas, después de lo cual el ritmo de destrucción equipara el ritmo de producción y por lo tanto su abundancia alcanza una meseta.[14]
La disposición de colágeno es importante porque el mismo aumenta la resistencia de la herida. En ausencia de colágeno lo único que mantiene a la herida cerrada es el coágulo de fibrina-fibronectina, que no provee demasiada resistencia frente a heridas traumáticas.[14] Además las células responsables de la inflamación, la angiogénesis y la construcción del tejido conectivo se adhieren, crecen y diferencian sobre la matriz de colágeno colocada por los fibroblastos.[27]
Aun cuando los fibroblastos producen nuevo colágeno, las colagenasas y otros factores lo degradan. Esta homeostasis marca el comienzo de la fase de maduración. Gradualmente termina la granulación y la cantidad de fibroblastos en la herida disminuye una vez que han cumplido con su misión.[28] Al final de la fase granular los fibroblastos comienzan a sufrir apoptosis, con lo que el tejido granular se transforma de un medio que es rico en células a uno que se compone principalmente de colágeno.[2]
Epitelización
La creación de tejido granular en una herida abierta permite que se desarrolle la fase de reepitelialización, durante la cual las células epiteliales migran a través del nuevo tejido para crear una barrera entre la herida y el medio ambiente.[25] Queratinocitos basales provenientes de los márgenes de la herida y apéndices dérmicos tales como folículos pilosos, glándulas sudoríparas y glándulas sebáceas son las principales células responsables de la fase de epitelialización de la cicatrización de la herida.[28] Las mismas avanzan formando una cubierta sobre el sitio de la herida y se desplazan desde los bordes hacia el centro de la herida.
Los queratinocitos migran primero para después proliferar.[29] La migración puede comenzar unos pocas horas luego de producida la herida. Sin embargo, las células epiteliales necesitan de un tejido viable para poder migrar a través del mismo, por lo que si la herida es profunda primero debe ser rellenada con tejido granular.[30] Por ello el tiempo para que comience la migración es variable y la migración puede recién comenzar un día después de producida la herida.[31] Las células de los márgenes de la herida proliferan durante el segundo a tercer día de producida la herida; esta es una manera de aumentar las células disponibles para la migración.[21]
Si la membrana basal no ha sido dañada, las células epiteliales son renovadas al cabo de tres días mediante división y migración hacia la superficie de células desde la capa basal de forma similar a lo que sucede en zonas de la piel que no han sufrido daño.[25] Sin embargo, si la membrana basal está dañada en la zona de la herida, la reepitelización solo se produce desde los márgenes de la herida y desde apéndices de la piel tales como folículos pilosos y glándulas sebáceas y sudoríparas que penetran la dermis y que poseen queratinocitos viables.[21] Si la herida es demasiado profunda los apéndices de la piel pueden también estar dañados y la migración desde los laterales de la herida se ve impedida.[30]
La migración de queratinocitos sobre la zona de la herida es estimulada por la ausencia de inhibición de contacto y por eleméntos químicos tales como el óxido nítrico.[32] Antes de comenzar a migrar las células deben disolver sus desmosomas y hemidesmosomas, los cuales normalmente anclan las células mediante filamentos intermedios de su citoesqueleto a otras células y al ECM.[17] Las proteínas receptoras de transmembrana llamadas integrinas, que están basadas en glicoproteínas y normalmente anclan las células a la membrana basal mediante su citoesqueleto, son liberadas de los filamentos intermedios de las células y se reconfiguran en filamentos de actina que sirven como vínculos a la ECM mediante pseudópodos durante la migración.[17] Por lo tanto los queratinocitos se desprenden de la membrana basal y pueden penetrar en la base de la herida.[23]
Antes de comenzar su migración los queratinocitos modifican su forma, tornándose más alargados y planos y extendiendo procesos celulares como los lamelipodia y otros procesos amplios que parecen ruffles.[19] Los filamentos de actina y pseudópodos form.[23] Durante la migración, las integrinas en el pseudópodo se toman de la ECM, y los filamentos de actina que se proyectan arrastran a la célula.[17] La interacción con las moléculas en el ECM mediante las integrinas promueve la formación de los filamentos de actina, lamelipodia y filopodia.[17]
Las células epiteliales se montan unas sobre otras para migrar.[28] Esta capa de células epiteliales que crece, a menudo es llamada la lengua epitelial.[29] Las primeras células que se adhieren a la membrana basal forman la capa basal. Estas células basales continúan su migración a través de la herida, y otras células epiteliales se deslizan sobre ellas.[29] Cuanto más rápido se produzca esta migración, tanto menor será la cicatriz que quede.[33]
La fibrina, el colágeno y la fibronectina que se encuentran en el ECM pueden inducir a las células a dividirse y a migrar. En la misma forma que los fibroblastos, los queratinocitos que migran utilizan la fibronectina entrelazada con fibrina que fue depositada en la inflamación como un sitio para sujetarse para poder reptar.[19][20][28]
En la medida que los queratinocitos migran se desplazan sobre el tejido granular, pero por debajo de la costra (si es que se formó una), separándola del tejido en su base.[28][31] Las células epiteliales poseen la habilidad de fagocitar residuos tales como tejido muerto y material bacteriano que si no obstruirían su paso. Debido a que deben disolver toda costra que se forme, la migración de queratinocitos es promovida por un medio húmedo, dado que un medio seco induce la formación de una costra más grande y más dura.[20][25][28][34] Para poder desplazarse a través del tejido, los queratinocitos deben disolver el coágulo, los residuos y partes del ECM para poder proseguir su viaje.[31][35] Ellos secretan un activador plasminogénico, el cual activa la plasmina para poder disolver el scab. Las células solo pueden migrar sobre tejido vivo por lo que deben secretar colagenasas y proteasas tales como metaloproteinasas matriciales (MMPs) para disolver las zonas dañadas del ECM que se encuentran en su camino, particularmente en la zona del frente de la placa migratoria.[28][31] Los queratinocitos también disuelven la membrana basal utilizano el nuevo ECM depositado por los fibroblastos para desplazarse.[17]
Mientras que los queratinocitos continúan migrando, se deben crear nuevas células epiteliales en los bordes de la herida para reemplazarlos y proveer más células a la capa que avanza.[20] La proliferación por detrás de los queratinocitos en migración por lo general comienza unos pocos días después de producida la herida[30] y tiene lugar a una velocidad que es 17 veces mayor en esta etapa de epitelialización que en tejidos normales.[20] Hasta que toda la zona de la herida es recubierta las únicas células epiteliales que proliferan son las de los bordes de la herida.[29]
Los factores de crecimiento estimulados por las integrinas y los MMPs, hacen que las células proliferen en los bordes de la herida. Los propios queratinocitos también producen y secretan factores, incluidos factores de crecimiento y proteínas de la membrana basal que ayudan tanto en la epitelialización como en otras fases del curado.[36]
Los queratinocitos continúan migrando a través de la herida hasta que la placa de células que avanza desde cada borde de la herida se encuentran en el centro, momento en el cual la inhibición por contacto hace que cese su migración.[19] Al terminar su migración, los queratinocitos secretan las proteínas que forman la nueva membrana basal.[19] Las células revierten los cambios morfológicos que sufrieron para comenzar su migración; restablecen sus desmosomas y hemidesmosomas y se fijan nuevamente a la membrana basal.[17] Las células basales comienzan a dividirse y diferenciarse de la misma manera en que lo hacen en la piel normal para restablecer la capa que normalmente se encuentra en la piel re-epitelializada.[19]
Contracción
Aproximadamente una semana luego de producida la herida, los fibroblastos se han diferenciado en miofibroblastos y la herida comienza a contraerse[37] En heridas profundas, la contracción alcanza su máximo de 5 a 15 días luego de producida la herida.[25] La contracción puede durar varias semanas y continúa aún después que la herida se ha reepitelializado por completo.[30][2] Si la contracción continúa por demasiado tiempo, puede producir desfiguración y pérdida de función.[38]
La finalidad de la contracción es disminuir el tamaño de la herida. Una herida grande puede reducir su tamaño entre un 40% a un 80% luego de la contracción.[19][28] Las heridas pueden contraerse a una velocidad de 0.75 mm por día, dependiendo de cuán flojo se encuentre el tejido.[25] La contracción por lo general no se produce de manera simétrica; la mayoría de las heridas poseen un 'eje de contracción' que posibilita una mejor organización y alineación de las células con el colágeno.[37]
Inicialmente, la contracción tiene lugar sin participación de los miofibroblastos.[39] Posteriormente, los fibroblastos, que han sido estimulados por factores de crecimiento, se diferencian en miofibroblastos. Los miofibroblastos, que son similares a las células de los músculos lisos, son los que realizan la contracción.[39] Los miofibroblastos contienen el mismo tipo de actina que existe en las células de los músculos lisos.[38]
Los miofibroblastos son atraídos por la fibronectina y factores de crecimiento y se desplazan mediante la fibronectina conectada a la fibrina en la ECM provisoria de manera de alcanzar los bordes de la herida.[20] Ellos establecen conexiones al ECM en los bordes de la herida, y se conectan unos con otros y a los bordes de la herida mediante desmosomas. También, mediante un enganche llamado fibronexus, la actina en los miofibroblastos es interconectada a través de la membrana de las células a moléculas como la fibronectina y el colágeno en la matriz extracelular.[39] Los miofibroblastos tienen numerosos enganches o adhesiones, que les permiten tirar de la ECM al contraerse, y reducir el tamaño de la herida.[38] En esta etapa de la contracción, el cierre de la herida ocurre más rápidamente que en la primera que no era producida por los miofibroblastos.[39]
Al contraerse la actina en los miofibroblastos, los bordes de la herida son juntados. Los fibroblastos depositan colágeno para reforzar la herida al contraerse los miofibroblastos[2] la etapa de contracción de la proliferación finaliza cuando los miofibroblastos detienen su contracción y se produce apoptosis.[38] La ruptura de la matriz provisoria conduce a una disminución en la concentración del ácido hialurónico y un incremento del sulfato de condroitina, que gradualmente conduce a los fibroblastos a detener su migración y proliferación.[13] Estos eventos marcan el comienzo de la etapa de maduración en la cicatrización de la herida.
Fase de maduración y remodelación
Cuando se igualan los niveles de producción y degradación de colágeno, se dice que ha comenzado la fase de reparación del tejido.[14] La fase de maduración puede durar un año o más, dependiendo del tamaño de la herida y si inicialmente se la cerró o se la dejó abierta.[21] Durante la maduración, se degrada el colágeno de tipo III, que era el que prevalecía durante la proliferación, y en su lugar se deposita el colágeno de tipo I que es más resistente.[11] Las fibras de colágeno que inicialmente se encuentran desorganizadas son interconectadas, ordenadas y alineadas a lo largo de líneas de tensión.[19] En la medida que la fase progresa, se incrementa la resistencia a la tracción de la herida, la resistencia alcanza un valor del 50% del de un tejido normal unos tres meses luego de ocurrida la herida y eventualmente alcanzando un 80% de la resistencia del tejido normal.[21] Dado que se reduce la actividad en la zona de la herida, la cicatriz pierde su apariencia eritematosa ya que los vasos sanguíneos que dejan de ser necesarios son eliminados mediante apoptosis.[14]
Las fases de cicatrización de una herida progresan normalmente en una forma predecible en el tiempo; si así no lo hicieran, el proceso de cicatrización puede evolucionar en forma indebida a una herida crónica tales como una úlcera venosa o una cicatriz patológica como por ejemplo una lesión queloide.[5][40][41]
Referencias
- Lara García T. Cicatrización de heridas cutáneas. Asociación de Clínica Estética. Asociación Médica Argentina; 2002.
- Stadelmann W.K., Digenis A.G. and Tobin G.R. (1998). Physiology and healing dynamics of chronic cutaneous wounds. The American Journal of Surgery 176 (2): 26S-38S. PMID 9777970.
- Iba Y., Shibata A., Kato M., and Masukawa T. (2004). Possible involvement of mast cells in collagen remodeling in the late phase of cutaneous wound healing in mice. International Immunopharmacology 4 (14): 1873-1880. PMID 15531302.
- Quinn, J.V. (1998). Tissue Adhesives in Wound Care. Hamilton, Ont. B.C. Decker, Inc. Electronic book.
- Midwood K.S., Williams L.V., and Schwarzbauer J.E. 2004. Tissue repair and the dynamics of the extracellular matrix. The International Journal of Biochemistry & Cell Biology 36 (6): 1031-1037. PMID 15094118.
- Chang H.Y., Sneddon J.B., Alizadeh A.A., Sood R., West R.B., Montgomery K., Chi J.T., van de Rijn M, Botstein D., Brown P.O. (2004). Gene Expression Signature of Fibroblast Serum Response Predicts Human Cancer Progression: Similarities between Tumors and Wounds. Public Library of Science 2 (2). PMID 14737219. Consultado el 20 de enero de 2008.
- Garg, H.G. (2000). Scarless Wound Healing. New York Marcel Dekker, Inc. Electronic book.
- Enoch, S. Price, P. (2004). Cellular, molecular and biochemical differences in the pathophysiology of healing between acute wounds, chronic wounds and wounds in the elderly. Worldwidewounds.com.
- Rosenberg L., de la Torre J. (2006). Wound Healing, Growth Factors. Emedicine.com. Accessed January 20, 2008.
- Sandeman S.R., Allen M.C., Liu C., Faragher R.G.A., Lloyd A.W. (2000). Human keratocyte migration into collagen gels declines with in vitro ageing. Mechanisms of Ageing and Development 119 (3): 149-157. PMID 11080534.
- Dealey C. (1999). The care of wounds: A guide for nurses. Oxford ; Malden, Mass. Blackwell Science. Electronic book.
- Theoret C.L. (2004). Update on wound repair. Clinical Techniques in Equine Practice 3 (2): 110-122.
- de la Torre J., Sholar A. (2006). Wound healing: Chronic wounds. Emedicine.com. Accessed January 20, 2008.
- Greenhalgh D.G. (1998). The role of apoptosis in wound healing. The International Journal of Biochemistry & Cell Biology 30 (9): 1019-1030. PMID 9785465.
- Muller M.J., Hollyoak M.A., Moaveni Z., La T., Brown H., Herndon D.N., Heggers J.P. 2003. Retardation of wound healing by silver sulfadiazine is reversed by Aloe vera and nystatin. Burns 29 (8): 834-836. PMID 14636760.
- Martin P. and Leibovich. (2005). Inflammatory cells during wound repair: the good, the bad and the ugly. Trends in Cell Biology 15 (11): 599-607. PMID 16202600.
- Santoro M.M. and Gaudino G. (2005). Cellular and molecular facets of keratinocyte reepithelization during wound healing. Experimental Cell Research 304 (1): 274-286. PMID 15707592.
- Expert Reviews in Molecular Medicine. (2003). The phases of cutaneous wound healing. 5: 1. Cambridge University Press. Accessed January 20, 2008.
- Lorenz H.P. and Longaker M.T. (2003). Wounds: Biology, Pathology, and Management. Stanford University Medical Center. Accessed January 20, 2008.
- Deodhar A.K., Rana R.E. (1997). Surgical physiology of wound healing: a review. Journal of Postgraduate Medicine 43 (2): 52-56. PMID 10740722. Accessed January 20, 2008.
- Mercandetti M., Cohen A.J. (2005). Wound Healing: Healing and Repair. Emedicine.com. Accessed January 20, 2008.
- Stashak T.S., Farstvedt E., Othic A. (2004). Update on wound dressings: Indications and best use. Clinical Techniques in Equine Practice 3 (2): 148-163.
- Falanga V. (2005). Wound Healing. American Academy of Dermatology (AAD).
- Kuwahara R.T. and Rasberry R. 2007. Chemical Peels. Emedicine.com. Accessed September 15, 2007.
- Romo T. and Pearson J.M. 2005. Wound Healing, Skin. Emedicine.com. Accessed December 27, 2006.
- Lansdown A.B.G., Sampson B., and Rowe A. 2001. Experimental observations in the rat on the influence of cadmium on skin wound repair. International Journal of Experimental Pathology, 82(1): 35-41. PMID 11422539.
- Ruszczak Z. 2003. Effect of collagen matrices on dermal wound healing. Advanced Drug Delivery Reviews, 55(12): 1595-1611. PMID 14623403
- DiPietro L.A. and Burns A.L., Eds. 2003. Wound Healing: Methods and Protocols. Methods in Molecular Medicine. Totowa, N.J. Humana Press. Electronic book.
- Bartkova J., Grøn B., Dabelsteen E., and Bartek J. 2003. Cell-cycle regulatory proteins in human wound healing. Archives of Oral Biology, 48(2): 125-132. PMID 12642231.
- Mulvaney M. and Harrington A. 1994. Chapter 7: Cutaneous trauma and its treatment. In, Textbook of Military Medicine: Military Dermatology. Office of the Surgeon General, Department of the Army. Virtual Naval Hospital Project. Accessed through web archive on September 15, 2007.
- Larjava H., Koivisto L., and Hakkinen L. 2002. Chapter 3: Keratinocyte Interactions with Fibronectin During Wound Healing. In, Heino, J. and Kahari, V.M. Cell Invasion. Medical Intelligence Unit ; 33. Georgetown, Tex., Austin, Tex Landes Bioscience, Inc. Electronic book.
- Witte M.B. and Barbul A. 2002. Role of nitric oxide in wound repair. The American Journal of Surgery, 183(4): 406-412. PMID 11975928.
- Son H.J. Bae H.C., Kim H.J., Lee D.H., Han D.W., and Park J.C. 2005. Effects of β-glucan on proliferation and migration of fibroblasts. Current Applied Physics, 5(5): 468-471.
- Falanga V. 2004. The chronic wound: impaired healing and solutions in the context of wound bed preparation. Blood Cells, Molecules, and Diseases, 32(1): 88-94. PMID 14757419.
- Etscheid M., Beer N., and Dodt J. 2005. The hyaluronan-binding protease upregulates ERK1/2 and PI3K/Akt signalling pathways in fibroblasts and stimulates cell proliferation and migration. Cellular Signalling, 17(12): 1486-1494. PMID 16153533.
- Bayram Y., Deveci M., Imirzalioglu N., Soysal Y., and Sengezer M. 2005. The cell based dressing with living allogenic keratinocytes in the treatment of foot ulcers: a case study. British Journal of Plastic Surgery, 58(7): 988-996. PMID 16040019. Accessed September 15, 2007.
- Eichler M.J. and Carlson M.A. 2005. Modeling dermal granulation tissue with the linear fibroblast-populated collagen matrix: A comparison with the round matrix model. Journal of Dermatological Science, 41(2): 97-108. PMID 16226016. Accessed September 15, 2007.
- Hinz B. 2005. Masters and servants of the force: The role of matrix adhesions in myofibroblast force perception and transmission. European Journal of Cell Biology 85(3-4): 175-181. PMID 16546559. Accessed September 15, 2007.
- Mirastschijski U., Haaksma C.J., Tomasek J.J., and Ågren M.S. 2004. Matrix metalloproteinase inhibitor GM 6001 attenuates keratinocyte migration, contraction and myofibroblast formation in skin wounds. Experimental Cell Research, 299(2): 465-475. PMID 15350544.
- O'Leary, R., Wood, E.J., and Guillou P.J. 2002. Pathological scarring: strategic interventions. European Journal of Surgery, 168(10):523-534. PMID 12666691.
- Desmouliere, A., Chaponnier, C., and Gabbiani, G. 2005. Tissue repair, contraction, and the myofibroblast. Wound Repair and Regeneration, 3(1):7-12. PMID 15659031.
Bibliografía
- Bauer SM, Bauer RJ, Liu ZJ, Chen H, Goldstein L, Velázquez OC. Vascular endothelial growth factor-C promotes vasculogenesis, angiogenesis, and collagen constriction in three-dimensional collagen gels. Journal of Vascular Surgery. 2005; 41(4): 699-707.
- Brain SD. Sensory neuropeptides: their role in inflammation and wound healing. Immunopharmacology. 1997; 37 (2-3):133-152.
- Ingber D. How wounds heal and tumors form Children's Hospital Boston research department.
- Losardo RJ. Tabaquismo crónico y cirugía plástica. Revista de la Asociación Médica Argentina. 2017; 130 (1):34-36.
- Mustoe T. 2005. Dermal ulcer healing: Advances in understanding. Presented at meeting: Tissue repair and ulcer/wound healing: molecular mechanisms, therapeutic targets and future directions. Paris, France, March 17-18, 2005. Consultado el 31 de diciembre de 2006.
- Revis D.R. and Seagel M.B. 2006. Skin, Grafts. Emedicine.com. Consultado el 31 de diciembre de 2006.
- Stillman R.M. 2006. Wound Care. Emedicine.com. Consultado el 31 de diciembre de 2006.
- Sturla F.M et al. 2013. La curación de las heridas de gran superficie y la medicina regenerativa. Revista de la Asociación Médica Argentina, Vol. 126, N.º 4, Pág. 12-23.
- Sturla F.M et al. 2018. La matriz extracelular en la curación de las heridas cutáneas. Aspectos físicos, químicos y biológicos.. Revista de la Asociación Médica Argentina, Vol. 131, N.º 2, Pág. 7-26.
- Wilhelmi B.J. 2006. Wound Healing, widened and hypertrophic scars. Emedicine.com. Consultado el 31 de diciembre de 2006.
Enlaces externos
- BioTherapeutics Education and Research Foundation
- Journal of Burns and Wounds
- suppliers of Medical Maggots
- Maggot Therapy Project
- Cicatrización en fumadores
 Datos: Q1509074
Datos: Q1509074 Multimedia: Wound healing / Q1509074
Multimedia: Wound healing / Q1509074