Catalyse
En chimie, la catalyse (du grec ancien : κατάλυσις / katálusis, « détacher »)[1] se réfère à l'accélération ou la réorientation de la cinétique de réaction au moyen d'un catalyseur, et dans certains cas à la sélectivité pour diriger la réaction dans un sens privilégié (réaction concurrente, production d'un produit plutôt qu'un autre)[2]. Le catalyseur est utilisé en quantité beaucoup plus faible que les produits réactifs. Il n'apparait pas en général dans le bilan de réaction, donc pas dans son équation globale. Cependant les molécules du catalyseur participent à la réaction dans une étape, ce qui explique leur influence sur la vitesse de réaction, et ensuite elles sont régénérées dans une étape subséquente[3]. Le catalyseur reste parfois infimement mélangé au produit final.
Pour l’article homonyme, voir Pot catalytique.

La catalyse joue un rôle dans de très nombreux domaines. Depuis plus de cent ans, elle a des applications dans le domaine du chauffage (exemple : la lampe Berger) : des combustions complètes, à température plus basse (moins dangereuses), quasiment sans flamme, et avec beaucoup moins de résidus de combustion dangereux (monoxyde de carbone, oxyde d'azote). Plus de 80 % des réactions chimiques industrielles sont réalisées à l'aide de procédés catalytiques en réduisant considérablement leur coût. Par exemple en 2007, les ventes mondiales de catalyseurs du domaine de dépollution des gaz de moteurs se montaient à environ seize milliards de dollars.
La vision populaire de ce domaine des catalyses est négative : pollution, dispersions de poussières de métaux rares et dangereux, vols de catalyseurs automobiles. Ces domaines de la science sont mal connus. [réf. souhaitée]
En biologie, dans les cellules, les enzymes, très nombreuses, jouent ces rôles d'accélérateur, de catalyseurs, dans les processus biochimiques : métabolisme digestif, de la reproduction, de la transcription de l'information génétique, les sciences du génome, le yaourt, la pâte à pain, etc.
Typologie
Différents types de catalyse peuvent être distingués selon la nature du catalyseur :
- catalyse homogène, si le catalyseur et les réactifs ne forment qu'une seule phase (souvent liquide) ;
- catalyse hétérogène, si le catalyseur et les réactifs forment plusieurs phases (généralement un catalyseur solide pour des réactifs en phase gazeuse ou liquide) ;
- catalyse enzymatique, si le catalyseur est une enzyme, c'est-à-dire une protéine ; de nombreux caractères de la catalyse enzymatique (influence de la concentration du catalyseur, types de succession d'étapes, etc.) sont les mêmes que ceux de la catalyse homogène.
La catalyse peut être aussi classée en fonction du mécanisme mis en jeu :
- catalyse acido-basique (générale ou spécifique) ;
- catalyse d'oxydoréduction ;
- catalyse nucléophile ;
- catalyse par transfert de phase.
Un catalyseur ne modifie ni le sens d'évolution d'une transformation ni la composition du système à l'état final. Tout catalyseur d'une réaction dans le sens direct catalyse aussi la réaction en sens inverse. De ce fait, un catalyseur ne permet pas à des réactions thermodynamiquement peu déplacées de modifier leur taux d'avancement final.
Par exemple, la réaction
ne se produit pas en l'absence de catalyseur (à température ordinaire) et aucun catalyseur ne peut la faire se produire avec un rendement satisfaisant.
Cependant, comme un catalyseur peut modifier fortement la vitesse d'une réaction parmi un grand nombre de réactions concurrentes possibles, il peut favoriser une réaction qui parait ne pas exister en son absence. C'est le cas de l'oxydation de l'ammoniac par le dioxygène :
- en l'absence de catalyseur, la réaction (non intéressante d'un point de vue industriel) est essentiellement : 2NH3 + O2 → N2 + 3H2O
- en présence de platine comme catalyseur, il se forme surtout du monoxyde d'azote (précieux intermédiaire dans la production d'acide nitrique) : 2NH3 + O2 → 2NO + 3H2O


Le monoxyde d'azote n'apparaît pas de façon mesurable en l'absence de platine.
Ne sont pas des catalyseurs…
- La température, bien qu'elle augmente la vitesse de réaction. Le catalyseur est une espèce chimique, ce que n'est pas la température.
- La lumière peut initier certaines réactions photochimiques, mais ce n'est pas un catalyseur pour autant car la lumière n'est pas une espèce chimique. De plus, la lumière ne ressort pas indemne, puisqu'une part de son énergie sera absorbée, et sa longueur d'onde sera donc modifiée.
- Un amorceur, bien que ce soit une espèce chimique, qu'il augmente la vitesse, et qu'il n'apparaisse pas dans le bilan. En effet, l'amorceur est consommé en début de réaction. Par exemple, un amorceur radicalaire permet de produire les premiers radicaux qui permettent à une polymérisation radicalaire de se produire.
Histoire
Étapes de la découverte

Quelques étapes notables sur la découverte et la compréhension de la notion de catalyse sont listées ici par ordre chronologique. Les premières découvertes sont liées au domaine de la biocatalyse :
- 6 000 ans av. J.-C., procédés techniques de la fermentation alcoolique des sucres, utilisés par les Sumériens en Mésopotamie : production d'acide acétique de l'alcool au moyen d'enzymes.
- Accélération et orientation des fermentations du lait pour les fromages : les découvertes liées permettaient très rapidement de diminuer les risques de colonisation par les agents pathogènes et augmentaient nettement la durée de consommation des produits laitiers (à l'échelle du temps du produit de base, c'était une technique de toute première importance).
- Les processus de panification des farines permettant la réalisation en épaisseur de cuisson au four et une modification de la digestibilité du composant de base.
Après ces débuts, la découverte de toute une série de nouvelles réactions catalytiques a eu lieu dans les XVIIIe et début du XIXe siècle.
- Au milieu du XVIIIe siècle, le physicien britannique John Roebuck élabore un procédé permettant la formation d'acide sulfurique par oxydation du dioxyde de soufre par l'air ; la réaction est effectuée en présence d'un catalyseur, l'acide nitrosylsulfurique, dans une cuve en plomb[4].
- Vers 1780, multiples découvertes Antoine-Augustin Parmentier, pharmacien militaire, agronome, nutritionniste et hygiéniste français : catalyse acide du sucre.
- 1782, découverte de Carl Wilhelm Scheele, sur l'estérification catalysée par un acide des alcools.
- 1783, Joseph Priestley, transformation de l'éthanol à partir d'éthylène.
- 1794, Elizabeth Fulhame, décrit dans son Essay on Combustion l'importance de l'eau dans certaines réactions d’oxydation.
- Dès 1814, Constantin Kirchhoff rapporte l'hydrolyse de l'amidon catalysée par les acides.
- En 1817, Humphry Davy découvre que l'introduction de platine chaud dans un mélange d'air et de gaz issus du charbon conduit à chauffer à blanc le métal.
- En 1824, William Henry rapporte l'empoisonnement d'un catalyseur : l'éthylène inhibe la réaction entre l'hydrogène et l'oxygène sur du platine. Il remarque par ailleurs l'oxydation sélective dans la réaction entre l'oxygène et un mélange gazeux composé d'hydrogène, de monoxyde de carbone et de méthane.
- Le terme catalyse est introduit par Berzelius en 1835 (du grec καταλύειν, qui signifie dénouer) pour nommer la cause d'un groupe de réactions[5]. Cette idée n'eut pas que des adeptes et Liebig, par exemple, s'opposa, dans le cas de la fermentation, à la théorie de la catalyse avancée par Berzelius. Pour Liebig, la cause de la fermentation était due à la transmission de vibrations des particules de ferment[6].
- En 1845, William Grove montre qu'un filament de platine est également un catalyseur pour la décomposition de l'eau en hydrogène et oxygène.
- Dans les années 1850, Kekulé comprit que la loi d'action de masse et les forces catalytiques avaient de l'importance sur les réactions chimiques, et que les forces qui favorisaient l'association des molécules Aneinanderlagerung causaient également leur décomposition, et que des produits intermédiaires pouvaient être isolés[7]. Voir la représentation ci-contre.

- En 1871, le procédé Deacon (en), utilisé pour l'oxydation de l'acide chlorhydrique en chlore, est développé : il utilise un catalyseur à base de brique en argile imprégnée de sels de cuivre.
- Peu de temps après, en 1877, Clément Georges Lemoine démontre que la décomposition de l'iodure d'hydrogène en dihydrogène et diiode atteint le même point d'équilibre à 350 °C que la réaction soit menée avec ou sans catalyseur (platine).
- Cette propriété est confirmée deux ans plus tard par Claude Louis Berthollet avec l'estérification des acides organiques et l'hydrolyse des esters dont l'équilibre de réaction reste identique avec ou sans catalyseur.
- Vers 1901, Georg Bredig mettait en avant l'importance de l'empoisonnement d'un catalyseur[8].
La catalyse au développement de la chimie
Le début du XXe siècle marque une découverte qui continue d'avoir des répercussions de nos jours[9]. Wilhelm Normann réalise l'hydrogénation de l'acide oléique (acide cis-9-octadécènoïque C17H33COOH), liquide, en acide stéarique (acide octadécanoïque C17H35COOH), solide, sur du nickel finement divisé. Cette hydrogénation est encore largement utilisée au XXIe siècle dans de nombreux domaines (alimentation, pharmacie, savonnerie, parfumerie, peinture, etc.) et le nickel reste le catalyseur phare.
La synthèse de l'ammoniac (NH3) à partir du diazote et du dihydrogène est mise au point par Fritz Haber sous haute pression, à température moyenne et catalysée par le fer (Fe3O4 réduit). Cet ammoniac peut être oxydé en monoxyde d'azote par oxydation, catalysé cette fois par le platine, pour servir de base à la fabrication d'acide nitrique (HNO3). En 1923, BASF commande une usine de fabrication du méthanol à partir de monoxyde de carbone et d'hydrogène sur un catalyseur à base d'oxyde de zinc et d'oxyde de chrome. Durant la même période, le procédé Fischer-Tropsch permet d'obtenir des alcanes, des alcènes et des alcools à partir de monoxyde de carbone et d'hydrogène à l'aide de catalyseur à base de fer et de cobalt. L'oxydation catalytique du dioxyde de soufre en trioxyde de soufre sur l'oxyde de vanadium(V) (V2O5) permet la synthèse à grande échelle d'acide sulfurique.
À la fin des années 1930, le craquage catalytique apparaît, offrant la possibilité de rompre les liaisons C-C. Ce procédé Houdry, utilise un catalyseur à base d'argile de type montmorillonite traitée à l'acide, et permet de scinder les grosses molécules du pétrole, typiquement contenues dans les gasoils, en plus petites constituant les essences. Durant la même décennie, l'oxydation sélective de l'éthylène en oxyde d'éthylène sur un catalyseur à base d'argent est mise au point, développée et commercialisée par Union Carbide. Tous ces procédés permettent d'avoir accès à une échelle industrielle à des produits de base de la chimie, ouvrant ainsi la voie au développement de la chimie de base et de spécialité.
Juste après la Seconde Guerre mondiale, les Trente Glorieuses profiteront largement à la chimie avec un grand développement des procédés en tout genre pour des productions de plus en plus diversifiées. La catalyse sera un acteur important de ce développement. La polymérisation se développe grandement en profitant des molécules de base produites. Dans les années 1950, le polyéthylène, le polypropylène et le polybutadiène apparaissent grâce notamment au procédé de polymérisation coordinative Ziegler-Natta utilisant des catalyseurs à base de complexes organométalliques de titane et aluminium. Le traitement du pétrole s'affirme avec l'hydrodésulfuration sur des catalyseurs à base de sulfure de cobalt et de molybdène, l'hydrotraitement des naphtas sur des catalyseurs cobalt-molybdène déposés sur alumine.
Les années 1960 marquent l'apparition des zéolithes de synthèse actives et sélectives pour l'isomérisation des alcanes et le craquage catalytique. Dès lors, ces matériaux vont faire l'objet d'intenses études pour leurs propriétés catalytiques et les chercheurs mettent au point de nombreuses zéolithes aux propriétés adaptées selon les réactions à catalyser, mais aussi à la forme des molécules par le contrôle de la taille des canaux. Les réactions mises en jeu conduisent à des molécules de plus en plus diverses : l'ammoxydation du propylène sur des catalyseurs à base d'oxydes de bismuth et de molybdène conduit à la fabrication d'acrylonitrile, alors que l'oxychloration de l'éthylène sur des catalyseurs à base de chlorure de cuivre(II) conduit au chlorure de vinyle.
La décennie 1970 voit apparaître le pot catalytique à base de platine, rhodium et palladium, alors qu'ailleurs se développe industriellement la catalyse enzymatique avec l'immobilisation d'enzymes, permettant de produire des pénicillines semi-synthétiques ou l'isomérisation du glucose en fructose. La découverte des zéolithes synthétiques aboutit industriellement dans les années 1980, le procédé MTG (methanol to gasoline : « méthanol vers essence ») permet de fabriquer de l'essence à partir du méthanol grâce au zéolithe H-ZSM5, production de diesel à partir CO et H2 grâce à des catalyseurs à base de cobalt. La chimie fine n'est pas en reste avec la synthèse de la vitamine K4 à l'aide d'un catalyseur membranaire à base de platine.
La liste est encore très longue et les molécules de plus en plus élaborées et de nouvelles perspectives s'ouvrent dans les années 2010[10],[11],[12],[13],[14],[15],[16],[17] avec l'utilisation de catalyseurs à un seul atome, qui devraient encore améliorer la catalyse hétérogène, dont dans les domaines de la chimie et de l'énergie[18].
Considérations générales
Catalyseur
En chimie, un catalyseur est une substance qui augmente la vitesse d'une réaction chimique ; il participe à la réaction dans une étape, mais est régénéré dans une étape subséquente. Il ne fait donc pas partie des réactifs. S'il fait partie des produits, la réaction est dite autocatalysée. C'est le cas par exemple de la réaction d'équation
- 2 MnO4− + 5 H2C2O4 + 6 H+ → 2 Mn2+ + 10 CO2 + 8 H2O
pour laquelle les ions Mn2+ ont un rôle catalytique.
Lorsqu’un catalyseur est utilisé pour accélérer une transformation, on dit que celle-ci est catalysée. Les catalyseurs agissent seulement sur des produits prédéterminés. Si un catalyseur accélère la réaction, il est dit positif. S'il la ralentit, il est dit négatif[19].
Les catalyseurs sont largement utilisés dans l'industrie et en laboratoire parce qu'ils augmentent considérablement la production des produits tout en minimisant les coûts de production. Dans la nature et en biochimie, certaines protéines possèdent une activité catalytique. Il s'agit des enzymes.
Le catalyseur augmente la vitesse de réaction en introduisant de nouveaux chemins de réaction (mécanisme), et en abaissant son énergie d'activation, ou énergie libre de Gibbs d'activation. Ce faisant il permet d'augmenter la vitesse, ou d'abaisser la température de la réaction. Le catalyseur ne modifie pas l'énergie libre de Gibbs totale de la réaction qui est une fonction d'état du système et n'a donc aucun effet sur la constante d'équilibre.
En plus de modifier la vitesse de réaction, le choix d'un catalyseur peut reposer sur d'autres paramètres :
- la sélectivité : un catalyseur sélectif favorise la formation du produit désiré par rapport aux produits secondaires. Par exemple, quand on utilise l'argent métallique pour catalyser la réaction de formation de l'oxyde d'éthylène, à partir d'oxygène et d'éthylène, cette réaction est accompagnée par la formation plus favorable thermodynamiquement de CO2 et H2O ;
- la durée de vie : le catalyseur doit pouvoir demeurer intact après plusieurs cycles de réaction.
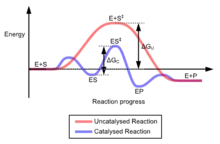
La succession des étapes conduisant à la formation d'un produit n'est pas la même en présence et en l'absence d'un catalyseur.
- En l'absence de catalyseur, l'énergie potentielle du système chimique prend une valeur importante rendant la réaction lente (ou par analogie la promenade épuisante).
- En revanche, la présence d'un catalyseur permet d'atteindre le même état final mais par une succession d'étapes dont aucune ne possède une énergie potentielle élevée.
Le chemin réactionnel est donc différent en présence et en absence du catalyseur ; le nombre d'étapes est généralement plus élevé avec catalyse que sans, mais toutes les étapes sont rapides.
Mécanisme type
Le catalyseur réagit généralement avec un ou plusieurs réactifs pour donner un intermédiaire, qui donne le produit de la réaction tout en régénérant le catalyseur. Par exemple, le bilan d'une réaction R → P, en présence d'un catalyseur (C) peut s'écrire :
- R + C → RC (1)
- RC → P + C (2)
- Ces deux étapes peuvent ou non être des équilibres chimiques.
Bien que le catalyseur soit consommé dans l'étape (1), il est régénéré par l'étape (2). La somme des deux étapes est donc identique au bilan annoncé :
- R → P
Cependant, le catalyseur parait généralement dans la loi de vitesse. Si l'étape cinétiquement déterminante au schéma ci-dessus est la première étape R + C → RC, la réaction catalysée sera du second ordre avec l'équation de vitesse v = kcat[R][C]. Mais le mécanisme catalysé a lieu en parallèle avec la réaction non catalysée. Si cette dernière est élémentaire, son équation de vitesse sera v = k0[R] et l'équation de vitesse globale sera v = k0[R] + kcat[R][C], que l'on peut écrire
- v = k[R], où k = k0 + kcat[C].
Ici le coefficient de vitesse (k) est la somme de deux termes. Le premier terme, normalement faible, représente la constante de vitesse de la réaction sans catalyseur. Le deuxième terme est proportionnel à la concentration du catalyseur, qui elle demeure constante lors de l'évolution d'une réaction dans le temps.
Catalyse et énergie de réaction
Un catalyseur fonctionne en permettant un mécanisme alternatif mettant en jeu différents états de transition et des énergies d'activation plus basses. Ainsi, dans le cas d'une réaction bimoléculaire simple de type A + B, l'état de transition est remplacé par un intermédiaire réactionnel de plus basse énergie, accompagné par deux états de transition, eux-mêmes de plus basse énergie. L'effet de ce changement est que plus de collisions moléculaires ont l'énergie nécessaire pour atteindre l'état de transition. Ainsi, un catalyseur permet d'effectuer des réactions qui, bien que thermodynamiquement faisables, étaient cinétiquement impossibles, ou nettement plus lentes. Un catalyseur abaisse donc l'énergie d'activation d'une réaction.
Un catalyseur ne peut pas rendre possible une réaction énergétiquement défavorable, pas plus qu'il ne peut déplacer l'équilibre final. La réaction et la réaction inverse sont également catalysées (principe de microréversibilité). L'enthalpie libre de la réaction est inchangée.
Classement selon la nature du catalyseur
Catalyse hétérogène
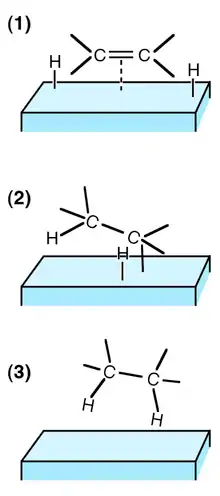
(1) Les réactifs sont adsorbés sur la surface du catalyseur et H2 est dissocié ;
(2) Un atome H se lie à l'un des atomes C. L'autre atome C est attaché à la surface ;
(3) Un 2e atome C se lie à un atome H. La molécule s'éloigne de la surface.
La catalyse est hétérogène quand le catalyseur et les réactifs ne sont pas dans la même phase. L'immense majorité des cas de catalyse hétérogène fait intervenir un catalyseur sous forme solide, les réactifs étant alors gazeux et/ou liquides. Les principales étapes du mécanisme sont décrites par la figure ci-contre.
Catalyse homogène
En catalyse homogène, les réactifs et le catalyseur sont présents dans la même phase. On retrouve beaucoup ce type de catalyse en chimie organique où de nombreuses réactions se déroulent avec des réactifs en solution, en présence d'ions H+, d'acides de Lewis, de complexes, etc., tous étant également solubles.
Catalyse enzymatique
Dans la biologie, les enzymes sont des catalyseurs des réactions métaboliques. Elles ont des structures basées sur des protéines. Les enzymes solubles peuvent être considérées comme intermédiaires entre les catalyseurs homogènes et hétérogènes; elles sont homogènes au niveau macroscopique mais au niveau moléculaire les réactions catalysées ont lieu sur la surface de l'enzyme comme pour la catalyse hétérogène. Les enzymes liés aux membranes biologiques par contre sont hétérogènes.
Classement selon le type de réaction activée
Catalyse acido-basique
Dans ces réactions, le catalyseur agit en tant qu'acide ou base. Cet acide ou cette base sont généralement des ions H+, HO−, des acides ou des bases de Lewis, ou encore des oxydes métalliques (Al2O3, V2O5, etc.). On distingue deux cas, selon que la réaction est accélérée par tous les acides (respectivement toutes les bases), ce qui s'appelle la catalyse générale, ou s'il faut un acide (ou une base) en particulier, ce qui s'appelle la catalyse spécifique.
Catalyse spécifique
Dans certains cas, un acide particulier sert de catalyseur. Le mécanisme passe alors par un mécanisme qui lui est propre, et qui serait différent pour un autre acide. C'est le cas de la réaction d'halogénation de la propanone[20] :
- CH3-CO-CH3 + X2 → CH3-CO-CH2X + HX
- X = I ou Br
Cette réaction est accélérée par H3O+ (ou par HO−). La constante de vitesse est de la forme
- k = k0 + k1[H3O+] + k2[HO−].
- avec k0, constante de vitesse de la réaction non catalysée. La valeur de k0 est très faible devant k1 et k2 (d'où l'effet notable de l'augmentation de la vitesse par les catalyseurs H3O+ et HO−).
L'ajout d'un acide faible ne modifie la vitesse que par la variation de la concentration [H3O+] qu'il permet, et non par la variation de sa propre concentration. Cela indique que c'est spécifiquement H3O+ le catalyseur, et non n'importe quel acide.
L'inversion du saccharose est également de type catalyse spécifique. Son équation est :
- Saccharose + eau → glucose + fructose
- soit C12H22O11 + H2O → C6H12O6 + C6H12O6
L'halogénation des nitroalcanes est un exemple de catalyse basique spécifique.
Catalyse générale
Pour qu'une catalyse soit acido-basique générale, il faut que des acides (ou des bases) faibles catalysent également la réaction. Cette catalyse doit dépendre de la concentration en acide faible, et pas seulement du fait que cet acide peut libérer des ions H+.
La constante de vitesse, en catalyse acide spécifique est donc de la forme :
- k = k0 + k1[H3O+] + k2[AH].
- où [AH] est la concentration en acide faible.
Pour montrer cette propriété de catalyse acide générale, il faut par exemple déterminer la dépendance de la vitesse (donc de k) en fonction de la quantité de AH ajouté, mais cela dans un milieu tamponné, afin que le terme k1[H3O+] soit maintenu constant[21].
Catalyse d'oxydoréduction
Des réactions d'oxydoréduction peuvent aussi être catalysées. Par exemple, la dismutation de l'eau oxygénée est catalysée par les ions Fe2+ ou Fe3+, l'hydrogénation des alcènes par le nickel de Raney, ou par ou métaux nobles supportés[22]. L'hydrogénation et l'hydrodésoxygénation sont largement étudiées en raison de leur pertinence pour la valorisation des composés biosourcés[23]. Les procédés oxydatifs sont utilisés pour la synthèse de produits chimiques tels que le formaldéhyde[24], acétaldéhyde[25], l'acide acrylique[26],[27], l'acide benzoïque[28]. Une telle catalyse met généralement en jeu un couple redox dont le potentiel sera compris entre le potentiel de l'oxydant et celui du réducteur.
- Catalyse nucléophile[réf. nécessaire]
Des réactions de substitutions nucléophiles peuvent être fortement accélérées en présence de traces d'autres nucléophiles. L'exemple classique est l'iodure de lithium. Dans ce sel, l'ion iodure est très peu lié au lithium, et est un nucléophile assez efficace. L'ion iodure est aussi un nucléofuge très efficace. Il sera donc déplacé par le nucléophile principal plus rapidement que ne se serait déroulée la réaction en absence de catalyseur.
- Catalyse par transfert de phase
Ici, l'idée est d'amener en contact des espèces se trouvant dans deux phases différentes. Ainsi, des substitutions nucléophiles, par exemple
- RCl + HO− → ROH + Cl−
seraient réalisables si la base HO− qui est en phase aqueuse et le substrat RCl qui en phase organique pouvaient se rencontrer. Une espèce chimique qui transporterait l'ion hydroxyde de la phase aqueuse à la phase organique, puis retransporterait le nucléofuge Cl− de la phase organique à la phase aqueuse sans se transformer lui-même serait un catalyseur, et dans ce cas, un catalyseur par transfert de phase. Une règle essentielle est que chaque phase doit respecter l'électroneutralité, si bien que si un cation change de phase, un anion doit changer en même temps (ou un cation doit passer en même temps dans l'autre direction).
Les cations ammonium substitués par des chaînes alkyle, par exemple (C4H9)N+, peuvent jouer un tel rôle catalytique. De par leur charge positive, ils peuvent être solvatés en phase aqueuse, et par leurs chaînes alkyle, ils peuvent l'être en phase organique. La première étape est dans ce cas, le transfert de (C4H9)N+ + HO− (espèce globalement neutre). La réaction de substitution peut avoir lieu en phase organique, et produire l'anion Cl−. L'espèce (C4H9)N+ + Cl− (toujours globalement neutre) peut passer en phase aqueuse. Le cation ammonium est alors à nouveau disponible pour un nouveau transfert d'ion HO−.
Les substitutions nucléophiles ne sont pas les seules à pouvoir mettre en jeu une catalyse à transfert de phase, par exemple l'oxydation du styrène par les ions permanganate[29], en présence de (C4H9)N+ + HSO4−.
- Autocatalyse
Dans certains cas, le catalyseur apparaît dans le bilan de la réaction, du côté des produits : la réaction est alors autocatalysée. L'effet d'une autocatalyse se traduit par une augmentation de la vitesse de réaction (alors que la vitesse diminue toujours lorsque la réaction avance) avant de diminuer. L'augmentation de la vitesse est due à l'augmentation de la concentration en catalyseur, et sa diminution à la disparition importante de ses réactifs.
Quelques exemples de réactions et de procédés

Un grand nombre de procédés chimiques comportent au moins une étape catalysée, que ce soit pour la fabrication de fibres synthétiques, de médicaments ou d'additifs alimentaires, sans compter toutes les réactions biologiques catalysées par les enzymes. Par ailleurs, en favorisant des réactions peu polluantes, la catalyse est un des piliers de la chimie verte. La suite de cette section fournit des exemples de réactions catalysées suivant les secteurs d'applications.
Catalyse dans la vie quotidienne

- Le chauffage individuel au gaz par catalyse n'est pas dangereux car il n'émet pratiquement pas de CO[30].
- Le pot catalytique est sans doute l'exemple le plus connu de catalyses.
- L'utilisation de la levure de boulanger pour la fabrication du pain.
Catalyse dans les grands procédés industriels

D'après[31].
Les procédés industriels qui utilisent des catalyseurs permettent d'économiser des produits chimiques (meilleurs rendements, synthèse en moins d'étapes), du temps (donc de l'argent) et de l'énergie en mettant en jeu des procédés à plus basse température. Quelques exemples de grands procédés industriels sont :
- synthèse de l'ammoniac NH3 à partir de ses éléments H2 et N2, catalysée par le fer (procédé Haber) ;
- synthèse de l'acide sulfurique par le procédé de contact (oxydation de SO2 en SO3 catalysé par V2O5) ;
- reformage en pétrochimie (conversion par le platine et l'alumine de molécules naphténiques en molécules aromatiques) ;
- craquage en pétrochimie pour transformer les coupes lourdes en petites molécules, grâce aux zéolithes.
Expériences utilisées pour l'enseignement de la catalyse
- Expérience de la lampe sans flamme (oxydation de l'éthanol par l'air en présence de cuivre)[32].
- Diverses décomposition de l'eau oxygénée par des ions fer, du platine, du dioxyde de manganèse, du sang, une rondelle de radis, etc.
- Réactions en chimie organique (estérification, hydrolyse, etc.) catalysées par un acide fort.
Catalyse amusante
- La cendre peut servir de catalyseur lors de la combustion du sucre. Ce dernier, chauffé au briquet, ne produit aucune flamme. Recouvert de cendre, il peut s'enflammer et brûle d'une flamme bleue.
- La lampe Berger élimine les mauvaises odeurs en diffusant une substance active dans l'air, par combustion catalytique de son substrat.[réf. souhaitée].
Notes et références
- Wilhelm Gemoll: Griechisch-Deutsches Schul- und Handwörterbuch, Munich / Vienne, 1965.
- L. Schuffenecker, G. Scacchi G., B. Proust, J.-F. Foucaut, L. Martel et M. Bouchy, Thermodynamique et cinétique chimiques, Éd. Tec & doc, coll. « info chimie », 1991, p. 351.
- (en) Laidler Keith J. et Meiser John H., Physical Chemistry (Benjamin/Cummings 1982), p. 424 (ISBN 0-8053-5682-7).
- (en) Benoît Join, « La catalyse », sur La Recherche, (consulté le ).
- J. R. Partington (en) (1989), A Short History of Chemistry, Éd. Dover, p. 196, 213.
- Ibid., p. 230.
- Ibid., p. 289.
- Ibid., p. 341.
- « Catalyse », sur physique-et-matiere.com (consulté le ).
- Sharon Mitchell et Javier Pérez-Ramírez, « Single atom catalysis: a decade of stunning progress and the promise for a bright future », Nature Communications, vol. 11, no 1, (ISSN 2041-1723, DOI 10.1038/s41467-020-18182-5, lire en ligne, consulté le )
- Abhaya K. Datye et Hua Guo, « Single atom catalysis poised to transition from an academic curiosity to an industrially relevant technology », Nature Communications, vol. 12, no 1, (ISSN 2041-1723, DOI 10.1038/s41467-021-21152-0, lire en ligne, consulté le )
- Hongzhou Yang, Lu Shang, Qinghua Zhang et Run Shi, « A universal ligand mediated method for large scale synthesis of transition metal single atom catalysts », Nature Communications, vol. 10, no 1, (ISSN 2041-1723, DOI 10.1038/s41467-019-12510-0, lire en ligne, consulté le )
- Huishan Shang, Xiangyi Zhou, Juncai Dong et Ang Li, « Engineering unsymmetrically coordinated Cu-S1N3 single atom sites with enhanced oxygen reduction activity », Nature Communications, vol. 11, no 1, (ISSN 2041-1723, DOI 10.1038/s41467-020-16848-8, lire en ligne, consulté le )
- Shi Fang, Xiaorong Zhu, Xiaokang Liu et Jian Gu, « Uncovering near-free platinum single-atom dynamics during electrochemical hydrogen evolution reaction », Nature Communications, vol. 11, no 1, (ISSN 2041-1723, DOI 10.1038/s41467-020-14848-2, lire en ligne, consulté le )
- Mengyao Ouyang, Konstantinos G. Papanikolaou, Alexey Boubnov et Adam S. Hoffman, « Directing reaction pathways via in situ control of active site geometries in PdAu single-atom alloy catalysts », Nature Communications, vol. 12, no 1, (ISSN 2041-1723, DOI 10.1038/s41467-021-21555-z, lire en ligne, consulté le )
- Hengpan Yang, Qing Lin, Chao Zhang et Xinyao Yu, « Carbon dioxide electroreduction on single-atom nickel decorated carbon membranes with industry compatible current densities », Nature Communications, vol. 11, no 1, (ISSN 2041-1723, DOI 10.1038/s41467-020-14402-0, lire en ligne, consulté le )
- Guodong Sun, Zhi-Jian Zhao, Rentao Mu et Shenjun Zha, « Breaking the scaling relationship via thermally stable Pt/Cu single atom alloys for catalytic dehydrogenation », Nature Communications, vol. 9, no 1, (ISSN 2041-1723, DOI 10.1038/s41467-018-06967-8, lire en ligne, consulté le )
- (en) « Single atom catalysts push the boundaries of heterogeneous catalysis », Nature Communications, vol. 12, no 1, , p. 5884, s41467–021–26130-0 (ISSN 2041-1723, PMID 34635653, PMCID PMC8505485, DOI 10.1038/s41467-021-26130-0, lire en ligne, consulté le )
- Association québécoise des utilisateurs de l'ordinateur au primaire-secondaire, « Facteurs qui influencent la vitesse d'une réaction », Association québécoise des utilisateurs de l'ordinateur au primaire-secondaire, (consulté le ).
- Schuffenecker L., Scacchi G., Proust B. Foucaut J.-F., Martel L. et Bouchy M. (1991), Thermodynamique et cinétique chimiques, Éd. Tec & doc, coll. « info chimie », p. 354.
- Schuffenecker L., Scacchi G., Proust B. Foucaut J.-F., Martel L. et Bouchy M. (1991), Thermodynamique et cinétique chimiques, Éd. Tec & doc, coll. « info chimie », p. 356.
- (en) Emőke Sikora, Adrienn Kiss, Zsuzsa H. Göndör et Péter Pekker, « Fine-tuning the catalytic activity by applying nitrogen-doped carbon nanotubes as catalyst supports for the hydrogenation of olefins », Reaction Kinetics, Mechanisms and Catalysis, (ISSN 1878-5204, DOI 10.1007/s11144-019-01705-7, lire en ligne, consulté le ).
- (en) Feras Alshehri, Clement Feral, Kathleen Kirkwood et S. David Jackson, « Low temperature hydrogenation and hydrodeoxygenation of oxygen-substituted aromatics over Rh/silica: part 1: phenol, anisole and 4-methoxyphenol », Reaction Kinetics, Mechanisms and Catalysis, vol. 128, no 1, , p. 23–40 (ISSN 1878-5204, DOI 10.1007/s11144-019-01630-9, lire en ligne, consulté le ).
- (en) Dan Chen, Jing Shi, Yanbin Yao et Shiwen Wang, « Enhanced catalytic activity towards formaldehyde oxidation over Ag catalysts supported on carbon nanotubes », Reaction Kinetics, Mechanisms and Catalysis, vol. 127, no 1, , p. 315–329 (ISSN 1878-5204, DOI 10.1007/s11144-019-01549-1, lire en ligne, consulté le ).
- (en) Mikhail V. Parfenov et Larisa V. Pirutko, « Oxidation of ethylene to acetaldehyde by N2O on Na-modified FeZSM-5 zeolite », Reaction Kinetics, Mechanisms and Catalysis, vol. 127, no 2, , p. 1025–1038 (ISSN 1878-5204, DOI 10.1007/s11144-019-01610-z, lire en ligne, consulté le ).
- (en) « Surface chemistry of phase-pure M1 MoVTeNb oxide during operation in selective oxidation of propane to acrylic acid », J. Catal., vol. 285, , p. 48-60 (lire en ligne).
- (en) « The reaction network in propane oxidation over phase-pure MoVTeNb M1 oxide catalysts », J. Catal., , p. 369-385 (lire en ligne).
- (en) « Multifunctionality of Crystalline MoV(TeNb) M1 Oxide Catalysts in Selective Oxidation of Propane and Benzyl Alcohol », ACS Catal., vol. 3, no 6, , p. 1103-1113 (lire en ligne).
- R. Barbe et J.-F. Le Maréchal (2007), La chimie expérimentale, t. 2, Dunod, Sciences sup, p. 39.
- Patrick Gélin, La combustion catalytique [PDF], IRCELyon (consulté le 27 novembre 2020).
- « 3 – Grands procédés industriels en catalyse hétérogène » [PDF], sur cours.espci.fr (consulté le ).
- « La lampe sans flamme », sur scienceamusante.net.
Voir aussi
Bibliographie
- Jacques Angenault, La Chimie - dictionnaire encyclopédique, Dunod, 1995 (ISBN 2-10-002497-3)
Articles connexes
- Réaction autocatalytique
- Pot catalytique
- Inhibiteur (chimie) • Poison de catalyseur
- Cycle carbone-azote-oxygène (phénomène nucléaire analogue à la catalyse et actif dans les étoiles)
Liens externes
- Ressource relative à la santé :
- Notices dans des dictionnaires ou encyclopédies généralistes :
- Patrick Gélin, La combustion catalytique [PDF], IRCELyon
- La catalyse, conférence filmée de Christian Minot, Universités de tous les savoirs
- Évolution de catalyseurs solides, SFRS/CERIMES (film pédagogique ancien, 1970)
 Portail de la chimie
Portail de la chimie