Cardiopathie congénitale
Les cardiopathies congénitales sont des malformations du cœur survenant au cours de sa formation, pendant la vie intra-utérine. Le terme « congénital » (littéralement « né avec ») est à distinguer du qualificatif « héréditaire » (« ce dont on hérite », ce qui est transmis par les ascendants). Une malformation congénitale, en particulier cardiaque, n'est pas obligatoirement héréditaire et peut correspondre à un accident isolé et qui ne se reproduira pas.
Ne doit pas être confondu avec L'Enfant bleu.
| Médicament | Prostaglandine E1 |
|---|---|
| Spécialité | Cardiologie |
| CIM-10 | Q20-Q26 |
|---|---|
| CIM-9 | 745-747 |
| OMIM | et 140500 234750 et 140500 |
| DiseasesDB | 17017 |
| MedlinePlus | 001114 |
| MeSH | D006330 |
![]() Mise en garde médicale
Mise en garde médicale
Elles sont extrêmement diverses, allant de la simple anomalie bénigne permettant la croissance de l'enfant sans aucun problème, jusqu'à la malformation grave, incompatible avec la survie du nouveau-né. Elles sont également de complexités diverses, rendant une évaluation obligatoire en milieu spécialisé. Certaines sont notamment à l'origine des « enfants bleus », c'est-à-dire qui présentent une cyanose à la naissance.
Jusqu'à quatorze fœtus sur mille sont porteurs d’une malformation cardiaque, faisant de ces dernières les malformations les plus fréquentes de l'enfant[2]. Le dépistage des cardiopathies congénitales est donc un objectif majeur de l'échographie morphologique du fœtus.
Le diagnostic permettra de faire des examens complémentaires, comme une amniocentèse pour la recherche d'une trisomie 21 ou une microdélétion 22q11.
Dans certains cas, on pourra proposer aux parents une interruption médicale de grossesse en cas de malformation incompatible avec la vie. La présence de certaines cardiopathies nécessite l'accouchement de la mère dans une maternité de niveau III pour une prise en charge optimale du nouveau-né.
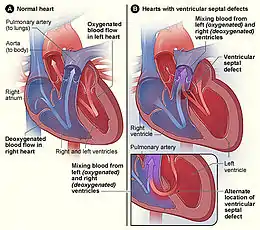
Les malformations cardiaques les plus fréquentes sont la communication inter-ventriculaire, la communication inter-auriculaire et la persistance du canal artériel. La communication inter-auriculaire par le foramen ovale et le passage par le canal artériel sont des éléments normaux de la circulation cardiaque du fœtus, mais qui disparaissent chez le nouveau-né normal.
Épidémiologie
La fréquence des cardiopathies congénitales est supérieure à 1 %. Si une mère a déjà accouché d’un enfant porteur d'une malformation cardiaque, le risque passe de 1 % à 5 % environ et il passe à 10 % si la mère a déjà accouché de deux enfants porteurs d'une malformation cardiaque. Une mère porteuse d'une malformation cardiaque a un risque augmenté de mettre au monde un enfant porteur d'une malformation cardiaque
Dans les syndromes d’origine génétique ayant une malformation cardiaque comme caractéristique, le risque de récurrence dépend du mode de transmission génétique de ce syndrome. Récemment, l'évaluation du risque de la trisomie 21 par mesure de la clarté nucale a permis de mettre en évidence que 10 % des embryons porteurs d'une clarté nucale anormale avec un caryotype normal ont une cardiopathie[réf. nécessaire].
Causes
Dans 85 % des cas, les cardiopathies congénitales sont d'origine inconnue[3]. Le diabète chez la mère (dit pré-gestationnel, c'est-à-dire connu avant le début de la grossesse) serait un facteur de risque[4].
Anomalies chromosomiques
Jusqu'à 40 % des fœtus ayant une malformation cardiaque ont des anomalies chromosomique comme la trisomie 21, la trisomie 13, la trisomie 18 ou d'autres anomalies chromosomiques.
Les malformations souvent associées à une anomalie chromosomique. Celles-ci incluent tétralogie de Fallot, hypoplasie du cœur gauche, canal atrioventriculaire et ventricule droit à double sortie. Les malformations rarement associées à une anomalie chromosomique. Celles-ci incluent transposition des gros vaisseaux et atrésie tricuspide. La fréquence des malformations cardiaques en fonction du type d'anomalie chromosomique impliquent 20 % des délétion 5p, 33 % des monosomies X, 50 % des trisomiques 21, 90 % des trisomiques 13 et 99 % des trisomiques 18[réf. souhaitée].
Le type de malformation cardiaque en fonction du type d'anomalie chromosomique : en cas de trisomie 21, le canal atrioventriculaire est l'anomalie la plus fréquente ; en cas de monosomie X, la coarctation de l'aorte est l'anomalie la plus fréquente.
Anomalies génétiques
Environ 5 % des malformations cardiaques sont en rapport avec une anomalie génétique. Près de 200 syndromes ont dans leur description une cardiopathie. La recherche sur Online Mendelian Inheritance in Man , site de référence sur les maladies génétiques retrouve 209 syndromes avec comme mot clé cardiopathies congénitales (congenital heart defect).
Certains syndromes n'entraînent pas des malformations cardiaques proprement dites, c'est-à-dire des anomalies anatomiques du cœur, mais sont responsables d'un mauvais fonctionnement cardiaque comme le syndrome de Romano-Ward et le syndrome de Wolff-Parkinson-White.
Les malformations cardiaques et maladies maternelles impliquent : diabète sucré, une maladie pouvant être responsable de malformations cardiaques, phénylcétonurie maternelle et lupus. Les malformations cardiaques et infection impliquent : la rubéole congénitale peut être responsable de rétrécissement de l'artère pulmonaire. Les malformations cardiaques et drogues impliquent : alcool, anti-inflammatoire, anti-convulsivant, barbiturique et lithium.
Embryologie
La formation du cœur commence 18 jours après la fécondation et se termine vers le 42e jour après la fécondation. Un des apports les plus intéressants de l'embryologie moderne est le fait que les cellules des crêtes neurales, à l'origine de la formation du cortex cérébral, migre dans le truncus arteriosus et le bulbis cordis, et participe aussi à la formation des gros vaisseaux cardiaques (artère pulmonaire et crosse de l'aorte).
Tube cardiaque
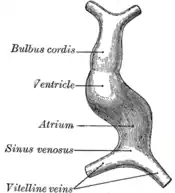
Le tube cardiaque se forme par fusion de la partie interne du mésoblaste ou splanchnopleure puis ce tube se renfle et forme des cavités alignées : oreillette primitive (atrium), le ventricule primitive (ventricle) et le truncus. Au niveau de l'oreillette primitive se trouve le sinus veineux et entre le truncus et le ventricule primitif, il existe un rétrécissement nommé conus (Dans le dessin à droite le troncus et le conus sont nommés bulbus cordis).
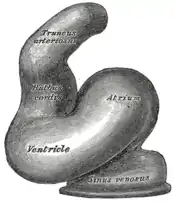
À 21 jours, le tube se courbe avec le truncus et le conus en haut et le sinus veineux en bas : les battements cardiaques apparaissent et la circulation commence ; il existe déjà une différenciation cellulaire en endocarde, myocarde et péricarde. Le tube cardiaque est entouré du mésocarde dans lequel des phénomènes d'apoptose apparaissent dans la partie postérieure du mésocarde aboutissant à la plicature du tube cardiaque au niveau du ventricule primitive avec majoration de la courbure et ascension relative du sinus veineux.
Cette courbure aboutit aussi à placer les futures valves auriculo-ventriculaires dans le même plan transversal.
Division des cavités cardiaques
La division de l'oreillette primitive et du ventricule primitive commence à 28 jours.
Au niveau de la partie postéro-supérieure de l'oreillete primitive apparaît un épaississement ou bourgeon qui s'étend vers le bas et l'avant formant le septum primum. Une partie de ce septum primum se développe plus lentement laissant persister un orifice : c'est l'ostium primum puis celui se comble et apparaît secondairement dans le septum primum une deuxième ouverture, remplaçant l'ostium primum qui se ferme, ou ostium secundum. L'oreillette primitive est maintenant divisée en oreillette gauche et droite.
Dans l'oreillette droite se forme une seconde cloison beaucoup plus épaisse que la première c'est le septum secundum dans lequel se trouve un orifice, le foramen ovale qui est situé à l'extrémité inférieur de l'ostium secundum. La cloison inter-auriculaire est donc formée à ce moment de deux parties : le septum primum, cloison très fine et le septum secundum musculaire et épais. Le sang passe de l'oreillette droite dans l'oreillette gauche par le foramen ovale du septum secundum puis l'ostium secundum du septum primum ; l'ensemble formant le canal de Botal qui a une forme en marche d'escalier du fait du non alignement des orifices.
Lors de la contraction des oreillettes, le sang passe par le canal de Botal de l'oreillette droite vers l'oreillette gauche en repoussant le septum primum formant une véritable valve; c'est la valve de Vieussen. Dans le même temps de la division des oreillettes la séparation des oreillettes et des ventricules s'opère.
Dépistage et diagnostic
Anténatal
Seule la recherche systématique soigneuse au cours de l'échographie morphologique, avec ou sans signe d'appel, peut permettre le dépistage des cardiopathies congénitales. Après localisation du dos et de la tête du fœtus, on localise le côté gauche du fœtus, et l'échographie de dépistage des cardiopathies congénitales peut commencer.
- Localisation du cœur en position intra thoracique
- Position du cœur à gauche dans le thorax
- Orientation ou cardie de la pointe du cœur vers la gauche
- Recherche des vaisseaux abdominaux (aorte et veine cave inférieure)
- Identification et évaluation des ventricules et des oreillettes
- Compte des vaisseaux sortant du cœur et identification de ces vaisseaux
- Étude de la crosse de l'aorte
- Recherche d'un retour veineux pulmonaire dans l'oreillette gauche par le doppler à codage couleur
Toute anomalie cardiaque fœtale doit faire chercher d'autres anomalies morphologiques associées, pouvant faire partie d'un syndrome. Toute anomalie cardiaque ou suspicion d'anomalie cardiaque doit être confirmée et évaluée par un cardiopédiatre qui permettra de confirmer ou d'infirmer le diagnostic et d'évaluer le pronostic (évolution spontanée) et les possibilités de traitement de la maladie.
Le tableau ci-dessous montre le taux de détection des cardiopathies congénitales graves sans une étude effectuée chez 30 149 fœtus correspondants au suivi de grossesse de à en Norvège à Trondheim avec suivi sur plusieurs années des enfants nés[5] : une malformation congénitale grave est une malformation nécessitant impérativement une intervention chirurgicale rapide. Une malformation cardiaque est dite complexe s'il existe une malformation impliquant un autre organe.
| Pathologie | Nombre | Fréquence de cette pathologie | Taux de détection | Taux de non détection | Note |
|---|---|---|---|---|---|
| Canal atrio-ventriculaire | 21 | 21 % | 71 % | 29 % | Dont 4 complexes |
| Transposition des gros vaisseaux | 17 | 12 % | 67 % | 33 % | Dont 7 complexes et 3 corrigées |
| Hypoplasie du cœur gauche | 10 | 11 % | 60 % | 40 % | Dont 2 complexes |
| Coarctation de l'aorte | 9 | 11 % | 60 % | 40 % | |
| Communication interventriculaire | 9 | 9 % | 33 % | 67 % | |
| Tétralogie de Fallot | 7 | 7 % | 43 % | 57 % | |
| Sténose pulmonaire | 4 | 4 % | 25 % | 75 % | |
| Atrésie pulmonaire | 3 | 3 % | 100 % | 0 % | |
| Communication inter auriculaire de type primaire | 3 | 3 % | 33 % | 67 % | |
| Ventricule unique | 3 | 3 % | 100 % | 0 % | |
| Ventricule droit à double sortie | 3 | 3 % | 67 % | 33 % | |
| Retour veineux pulmonaire anormal total | 2 | 2 % | 0 % | 100 % | |
| Atrésie de la tricuspide | 2 | 2 % | 50 % | 50 % | |
| Tronc artériel commun | 1 | 1 % | 100 % | 0 % | |
| Sténose aortique congénitale | 1 | 1 % | 100 % | 0 % | |
| Atrésie de la mitrale | 1 | 1 % | 100 % | 0 % | |
| Anomalie d'Ebstein | 1 | 1 % | 100 % | 0 % | |
| TOTAL | 97 | 100 % | 57 % | 43 % |
Postnatal
La cardiopathie est soupçonnée par la présence d'un souffle à l'auscultation cardiaque, d'une cyanose, d'une coloration bleutée de la peau. Mais près du quart des cardiopathies congénitales ne sont diagnostiquées qu'après la sortie du nouveau-né de la maternité[6].
Classifications
Plusieurs classifications sont utilisées pour classer les différentes cardiopathies :
- classification anatomique aussi appelée « segmentaire » ;
- classification selon le retentissement physiologique : les « non cyanogènes » et les « cyanogènes » ;
- la plus récente est une classification se basant sur les progrès des mécanismes embryologiques de la formation des cardiopathies.
Classification anatomique
Mise au point en 1964 par Stella Van Praagh cette classification permet de décrire toutes les cardiopathies congénitales à l'aide d'un système de lettre. Elle prend en compte le situs auriculaire (c’est-à-dire la position de l'oreillette droite dans le thorax : si l'oreillette droite est à droite on emploie la lettre D, à gauche la lettre L et en position indéterminée la lettre I), la courbure de la boucle ventriculaire (la courbe est dite D pour la courbe droite de la boucle ventriculaire positionnant le ventricule droit à droite du ventricule gauche ; la courbe est dite L pour la courbe gauche de la boucle ventriculaire positionnant le ventricule droit à gauche du ventricule gauche), et la position des gros vaisseaux (aorte et artère pulmonaire) soit le « Normo posé » Solitus (l'aorte est en arrière et à droite par rapport à l'artère pulmonaire ou Inversus) ; « Mal posé » avec emploi de la lettre D si la valve aortique est en avant et à droite par rapport à la valve pulmonaire, emploi de la lettre L si la valve aortique est en avant et à gauche par rapport à la valve pulmonaire et emploi de la lettre A ou « Antéro posé » la valve de l'aorte est en avant par rapport à la valve pulmonaire.
Classification physiologique
Les cardiopathies cyanogènes sont des malformations cardiaques où il y a un mélange de sang non oxygéné (bleu) avec du sang oxygéné (rouge). Une coloration bleu violacé de la face, des lèvres et des ongles apparaît, surtout lors de pleurs, due à une oxygénation insuffisante du sang d'où le nom de « maladie bleue ».
Par définition, elles nécessitent une communication entre le cœur droit et le cœur gauche. Or le régime de pression fait que, dans ce cas, le sang va du cœur gauche vers le cœur droit (pressions gauches supérieures aux pressions droites). Pour inverser le shunt, il faut inverser le gradient de pression, et par conséquent, augmenter celles des cavités droites. Le mécanisme le plus fréquent est un obstacle sur la voie d'éjection droite, comme c'est le cas dans la tétralogie de Fallot. Lorsque le mécanisme de l'inversion du shunt est une hypertension artérielle pulmonaire secondaire à la cardiopathie congénitale, il s'agit d'un syndrome d'Einsenmenger. Les cardiopathies cyanogènes sont donc plus complexes, associant au minimum une communication entre le cœur droit et le cœur gauche, et un obstacle à l'éjection sur le cœur droit.
Les cardiopathies non cyanogènes sont des malformations cardiaques dues à un shunt gauche-droit, à savoir une anomalie caractérisée par le mélange de sang oxygéné (plus clair) à du sang non oxygéné (plus foncé) (communication entre le cœur droit et le cœur gauche). Des malformations dues à un obstacle situé au sein du cœur ou dans un gros vaisseau (artère pulmonaire, aorte) peuvent également être mises en cause.
| Classification physiologique | |||||
|---|---|---|---|---|---|
| Cardiopathies cyanogènes | |||||
| Tétralogie de Fallot | |||||
| Sténose et atrésie pulmonaire (à condition d'avoir un shunt intra cardiaque) | |||||
| Transposition des gros vaisseaux | |||||
| Atrésie tricuspide | |||||
| Oreillette unique | |||||
| Retour veineux anormal total ou partiel | |||||
| Tronc artériel commun | |||||
| Cœur univentriculaire | |||||
| Maladie d'Ebstein | |||||
| Syndrome d'Einsenmenger | |||||
| Cardiopathies non cyanogènes | |||||
| Communication inter-auriculaire | |||||
| Communication inter-ventriculaire | |||||
| Canal atrio-ventriculaire | |||||
| Persistance du canal artériel | |||||
| Rétrécissement aortique | |||||
| Coarctation de l'aorte | |||||
| Sténose de l'artère pulmonaire | |||||
| Non compaction ventriculaire gauche | |||||
Anomalies associées
Elles dépendent du type de cardiopathie, certaines de ces dernières pouvant faire partie d'une maladie congénitale associant différentes anomalies de divers organes.
Les cardiopathies congénitales complexes sont cependant fréquemment associées à un retard de développement psychomoteur. Ces derniers ont été attribués, dans un premier temps, aux conséquences du bas débit cérébral imposé par la chirurgie correctrice, mais plusieurs arguments sont en faveur d'une origine plus précoce. Ainsi, près de la moitié de ces enfants ont des anomalies neurologiques avant toute cure chirurgicale[7]. De même, l'IRM des nouveau-nés et jeunes enfants, porteurs de cardiopathies congénitales complexes, semble montrer un taux élevé d'anomalies ressemblant à celles détectées chez les grands prématurés, faisant suggérer un défaut de maturation cérébrale pouvant être dû à une souffrance intra utérine secondaire aux problèmes cardiaques[8].
Voir aussi
Notes et références
- (en) JI Hoffman et S Kaplan, « The incidence of congenital heart disease », J. Am. Coll. Cardiol., vol. 39, no 12, , p. 1890–900 (PMID 12084585, DOI 10.1016/S0735-1097(02)01886-7)
- (en) E. Tegnander, W. Williams, O.J. Johansen, H.. K. Blaas, S.H. Eik-Nes Prenatal detection of heart defects in a non-selected population of 30 149 fetuses - detection rates and outcome Ultrasound in Obstetrics and Gynecology, Volume 27, Issue 3, Pages 252-265. Mars 2006
- .
- Øyen N, Diaz LJ, Leirgul E et al. Prepregnancy diabetes and offspring risk of congenital heart disease: A nationwide cohort study, Circulation, 2016;133:2243-2253
- (en) E. Tegnander, W. Williams, O.J. Johansen, H.. K. Blaas, S.H. Eik-Nes Prenatal detection of heart defects in a non-selected population of 30 149 fetuses - detection rates and outcome Ultrasound in Obstetrics and Gynecology, Volume 27, Issue 3, Pages 252-265, mars 2006
- Brown KL, Ridout DA, Hoskote A, Verhulst L, Ricci M, Bull C, Delayed diagnosis of congenital heart disease worsens preoperative condition and outcome of surgery in neonates, Heart, 2006;92:1298–1302
- Limperopoulos C, Majnemer A, Shevell MI et al. Neurologic status of newborns with congenital heart defects before open heart surgery, Pediatrics, 1999;103:402-408
- Miller SP, McQuillen PS, Hamrick S et al. Abnormal brain development in newborns with congenital heart disease, N Engl J Med, 2007;357:1928-1938
 Portail du handicap
Portail du handicap  Portail de la médecine
Portail de la médecine