Myasthénie
La myasthénie (du grec μύς « muscle » et ἀσθένεια « faiblesse »), en latin myasthenia gravis, est une maladie neuromusculaire auto-immune chronique, qui se manifeste à des degrés divers par une faiblesse et une fatigabilité excessive des muscles striés squelettiques, c'est-à-dire ceux qui sont activés volontairement. Elle doit son nom au neurologue allemand Friedrich Jolly (1844-1904) et possède également une dénomination éponymique : la maladie d'Erb-Goldflam, en l'honneur des deux cliniciens en ayant donné la description la plus complète. Elle est causée par des anticorps circulants qui, dans plus de trois cas sur quatre, ciblent les récepteurs de l'acétylcholine de la jonction neuromusculaire, inhibant ainsi l'effet excitateur de ce neurotransmetteur.
| Médicament | Pyridostigmine, néostigmine, ciclosporine, (RS)-cyclophosphamide, prednisone, azathioprine, rituximab, éculizumab, mycophénolate mofétil (d), prednisolone, tacrolimus, distigmine (d) et pyridostigmine |
|---|---|
| Spécialité | Neurologie |
| CIM-10 | G70.0 |
|---|---|
| CIM-9 | 358.0 |
| OMIM | 254200 |
| DiseasesDB | 8460 |
| MedlinePlus | 000712 |
| eMedicine |
1171206 emerg/325 (urgence), med/3260 (grossesse), oph/263 (œil) |
| MeSH | D009157 |
| Patient UK | Myasthenia-gravis-pro |
![]() Mise en garde médicale
Mise en garde médicale
Historique
C’est vraisemblablement Thomas Willis qui décrit le premier une patiente atteinte de myasthénie. Jolly nomme la maladie en inventant le terme de « myasthenia gravis pseudoparalytica » et met en évidence la détérioration électrophysiologique. Mary Walker propose en 1934 le traitement par la physostigmine[1]. Les anomalies du thymus en rapport avec la MG sont connues, et la thymectomie pratiquée, dès le début du XXe siècle, avant la compréhension de la physiopathologie et la mise en évidence de la genèse auto-immune.
Épidémiologie
Sa prévalence varie de 5 à 10 pour 100 000 habitants et son incidence entre 2 et 20 par année et par million d’habitants selon les séries. Pouvant survenir à tout âge, son pic d'incidence concerne la tranche d'âge des 20 à 30 ans. Le ratio de genre des malades, de 3 femmes pour 2 hommes, varie selon la tranche d'âge (2 femmes pour 1 homme pour les moins de 50 ans, 1 femme pour 1 homme pour les plus de 50 ans). Maladie essentiellement sporadique, les formes familiales sont exceptionnelles[2],[3],[4],[5],[6].
Physiopathologie

La physiopathologie supposée de la myasthénie consiste en une atteinte de protéines de la membrane post-synaptique de la jonction neuro-musculaire par des auto-anticorps circulants. Le principal anticorps identifié cible le récepteur à l'acétylcholine (un neurotransmetteur), se comportant comme un inhibiteur compétitif de l'acétylcholine et accélérant la dégradation du récepteur par modulation antigénique[7]. Il en résulte une augmentation du nombre nécessaire de quanta d'acétylcholine pour produire la dépolarisation de la membrane post-synaptique, entraînant un épuisement plus rapide et profond des réserves présynaptiques d'acétylcholine et se traduisant cliniquement par une fatigabilité musculaire.
À moyen terme, la présence de ces anticorps peut induire, via l'activation du système du complément, des altérations anatomiques de la membrane post-synaptique, diminuant encore son excitabilité. À ce stade, dans les formes les plus sévères, des déficits permanents peuvent s'installer.
À l'exception de la D-pénicillamine et de la greffe de moelle allogène, aucun agent exogène favorisant la survenue d'une myasthénie n'a été identifié. Il existe une légère prédisposition génétique, en particulier les groupes HLA B8 (en) et DR3 (en) avec le DR1 (en) plus spécifique d'une atteinte de myasthénie oculaire. De nombreux autres auto-anticorps ont récemment été identifiés comme pathogènes, tous ciblant un composant de la membrane post-synaptique : les anticorps anti-MuSK (muscle specific kinase), les anticorps anti-agrine, les anticorps anti-LRP4[8]. Le rôle du thymus dans la pathogénèse est démontré. Il est le siège de la maturation des lymphocytes T. Sous la stimulation de ces derniers, les lymphocytes B se transforment en plasmocytes sécrétant les anticorps[5],[7],[9],[10].
Manifestations cliniques
Les caractéristiques cliniques peuvent se décrire ainsi[5],[6],[11],[12],[13],[14] :
le symptôme cardinal et indispensable au diagnostic est la fatigabilité à l'effort. Les symptômes sont donc fluctuants, s'intensifiant à l'effort et s'améliorant au repos. Ils sont souvent plus marqués en fin de journée et peuvent s'aggraver dans des situations de stress : infection, intervention chirurgicale, chaleur[15]… Enfin, de ce fait, l'examen clinique peut être strictement normal ou peu perturbé, des tests d'efforts permettant alors de démasquer des anomalies.
La localisation musculaire et la symptomatologie en découlant sont variables :
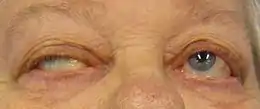
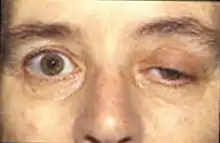
- dans 50 % des cas, la plainte initiale est un ptosis, une diplopie ou les deux à la fois. Ces symptômes sont souvent fluctuants, apparaissant ou s'aggravant généralement en fin de journée ou à la lecture ou lors de la conduite de véhicule. C'est la plainte la plus évocatrice du diagnostic et permettant le seul test diagnostique clinique spécifique. L'apparition d'une ptôse avec ophtalmoparésie (chute des paupières avec difficulté de convergence) à la fixation excentrée du regard prolongée quelques minutes chez un patient asymptomatique au préalable est en effet caractéristique. Chez un patient symptomatique, le test du glaçon (amélioration du ptosis après la pose de glace durant quelques minutes sur la paupière symptomatique) est moins spécifique mais très évocateur ;
- dans 20 % des cas, la plainte initiale concerne une fatigabilité des membres, le plus souvent dans ses parties proximales, pouvant égarer le diagnostic vers une myopathie. Des tests d'efforts répétés peuvent démasquer une fatigabilité segmentaires mais ne sont que peu spécifiques ;
- dans 10 % des cas, la plainte initiale consiste en une asthénie (fatigue généralisée, souvent confondue avec une véritable faiblesse musculaire) isolée, ce qui ne doit pas faire évoquer le diagnostic en première intention à moins qu'une véritable faiblesse musculaire ait été objectivée par un médecin et que la fatigabilité soit à l'avant-plan. Il s'agit de la plainte la plus aspécifique et les tests cliniques ne sont que de peu d'utilité ;
- assez rarement, les plaintes initiales consisteront en des troubles fluctuants de mastication, de la déglutition ou une faiblesse des extenseurs du cou ;
- l'observation d'une atteinte oculo-céphalique globale réalisant le « faciès myasthénique » est rare mais très évocatrice (ptôse palpébrale asymétrique, lèvre inférieure pendante, effacement des rides du front, éventuels troubles oculo-moteurs extrinsèques et faiblesse des extenseurs du cou). Le caractère fluctuant de ce faciès permet de le distinguer des nombreuses autres causes de diplégie faciale et d'affirmer le diagnostic.
Par définition, la symptomatologie est uniquement motrice et les plaintes sensitives absentes. Du fait d'efforts excessifs compensatoires, le patient peut cependant mettre en avant des plaintes sensitives, douloureuses (myalgies, arthralgies, paresthésies) égarant le diagnostic vers une pathologie rhumatismale, une myopathie, un trouble métabolique, une polyneuropathie ou une myélopathie.
À l'examen, les réflexes myotatiques sont normaux et une amyotrophie n'est observée que dans les formes sévères évoluées.
On distingue les formes généralisées des formes bulbaires et des formes oculaires pures. Diverses combinaisons peuvent se constituer (formes oculo--bulbaires). Les formes oculaires pures sont limitées à une atteinte de l'oculomotricité extrinsèque et à un ptosis . Dix à vingt pour cent (10 à 20 %) des formes oculaires resteront à ce stade, forme handicapante mais bénigne de la maladie, les autres évoluant vers une myasthénie généralisée.
Les formes bulbaires initiales (15 %) comportent une voix nasonnée avec difficulté articulatoire, une faiblesse masticatoire avec tendance à la chute de la mâchoire, une difficulté à la déglutition (dysphagie) avec des fausses routes alimentaires ou salivaires.
L'évolution de la maladie se fait typiquement par une succession d'aggravations et de rémissions, entrecoupées possiblement de véritables « crises myasthéniques » pour les formes généralisées. Ces crises, véritables urgences médicales, sont définies par une aggravation brutale avec insuffisance respiratoire aiguë, dyspnée avec encombrement, troubles de la déglutition avec risque de fausses routes ou faiblesse généralisée sévère avec impotence ne cédant pas au repos.
Le stade de gravité maximale est atteinte dans les trois ans chez plus des trois quarts des malades.
Dans 1 à 5 % des cas, la maladie se déclare par une crise myasthénique inaugurale. Le diagnostic est alors extrêmement difficile et doit être évoqué en cas de disproportion entre la pathologie causale présumée et la sévérité clinique ou en cas d'échecs répétés inexplicables à des tentatives de sevrage de ventilation mécanique, l'autre diagnostic à évoquer dans de telles situations étant une polyradiculo-névrite aiguë.
Classification clinique d'Osserman ou MGFA (Myasthenia Gravis Foundation of America)
Cette classification décrit plusieurs stades[16] :
- stade I : une faiblesse d'un muscle oculaire, possible ptosis. Pas d'autre faiblesse musculaire ;
- stade II : faiblesse des muscles oculaires de n'importe quelle sévérité, faiblesse légère des autres muscles :
- stade IIa : faiblesse prédominante aux muscles des membres ou au muscles du tronc,
- stade IIb : faiblesse prédominante aux muscles respiratoires ou troubles de la déglutition ;
- stade III : faiblesse des muscles oculaires de n'importe quelle sévérité, faiblesse modérée des autres muscles :
- stade IIIa : faiblesse prédominante aux muscles des membres ou au muscles du tronc,
- stade IIIb : Faiblesse prédominante aux muscles respiratoires ou troubles de la déglutition ;
- stade IV : faiblesse des muscles oculaires de n'importe quelle sévérité, faiblesse sévère des autres muscles :
- stade IVa : faiblesse prédominante aux muscles des membres ou au muscles du tronc,
- stade IVb : faiblesse prédominante aux muscles respiratoires ou troubles de la déglutition. Peut aussi inclure, une nécessité de sonde naso-gastrique sans intubation ;
- stade V : intubation nécessaire pour maintenir la respiration.
Formes particulières
Différentes formes de myasthénies méritent d'être individualisées de par leurs présentations et leurs prises en charge particulières[6].
Myasthénies fœtales et néo-natales auto-immunes
Le placenta est perméable aux anticorps. Dix à vingt pour cent (10 à 20 %) des enfants de mères myasthéniques présentent une myasthénie néonatale survenant dans les 3 premiers jours et régressant généralement en quelques semaines (maximum 3 mois). Les symptômes variables d'un enfant à l'autre comportent : une hypotonie, un cri faible, des troubles de la succion ou de la déglutition, un ptosis, une diplégie faciale et des troubles respiratoires. Le traitement repose sur une prise en charge symptomatique en unité de réanimation néo-natale jusqu'à résolution et en l'administration d'anti-cholinestérasiques. L'évolution est généralement favorable. Dans les formes les plus sévères peut être réalisée une exsanguinotransfusion.
Les formes fœtales sont plus rares et peuvent se traduire par des retards de croissance intra-utérine. Exceptionnellement, des manifestations cliniques sévères surviennent (hydramnios, arthrogrypose, mort in-utero).
Myasthénies infantiles et juvéniles auto-immunes
Myasthénie gravis débutant avant 15 ans, cette forme mérite d'être individualisée de par la distinction difficile avec les myasthénies congénitales non auto-immunes et la nécessité au vu de l'âge de restreindre au maximum l'usage des corticoïdes et immunosuppresseurs[17].
Myasthénies iatrogènes auto-immunes
Exceptionnelles, leur diagnostic est généralement évident sur base de l'histoire clinique. Elles impliquent les mêmes auto-anticorps et répondent aux mêmes traitements que la myasthénie classique. Elles sont cependant susceptibles de répondre à un traitement spécifique. Jusqu'ici, seules ont été démontrées :
- la myasthénie induite par la D-pénicillamine. Elle concerne 1 % des patients traités par D-pénicillamine. La guérison après arrêt de la D-pénicillamine dans les 6 mois est la règle ;
- la myasthénie induite par la transplantation de moelle allogénique avec maladie du greffon contre l'hôte.
Pathologies associées
Certaines pathologies, de par la fréquence de leur association à la myasthénie ou de par la gravité de leur pronostic spontané propre, sont à rechercher systématiquement[11],[12],[5],[13],[6],[14].
Tumeurs thymiques
Dans 10 à 15 % des cas de myasthénie, avec anticorps anti-récepteur à l’acétylcholine, un thymome est retrouvé. Les thymomes sont des tumeurs du médiastin antérieur, souvent bénignes, associées de manière variable à la myasthénie.
Un marqueur de l’existence de ces thymomes pour les patients de moins de 60 ans est l’anti-titine[18] L'indication chirurgicale s’impose. Elle permet de préciser si le thymome est bénin (2/3 cas) ou malin en fonction de son caractère invasif ou non.
Dans 50 à 70 % de la forme jeune de myasthénie avec anticorps anti-récepteur à l’acétylcholine, une hyperplasie thymique est retrouvée. Cette anomalie est toujours à considérer comme bénigne (ce qui n’exclut pas l’ablation du thymus).
Entre 15 et 20 % des patients présentant un thymome présentent également des signes cliniques de myasthénie, et 25 % des patients présentent des anticorps anti-récepteurs de l'acétyl-choline (anticorps anti-ACh-R) sans avoir de symptomatologie. Tout patient porteur d'un thymome présentant des signes de myasthénie a des anticorps positifs, et toute masse médiastinale asymptomatique associée à des anticorps anti-ACh-R est un thymome. Si l'association statistique suggère bien une corrélation entre les deux pathologies, le mécanisme exact de cause à effet demeure débattu et plusieurs hypothèses sont envisagées[19] :
- sélection de lymphocytes T défectueux au sein du thymome, responsables de l'hypersécrétion de l'auto-anticorps ;
- présentation défectueuse des auto-antigènes aux cellules thymiques, menant à la non élimination des lymphocytes T les reconnaissant ;
- présence de centres germinaux spécifiques au sein du parenchyme thymique produisant plus volontiers les auto-anticorps ;
- chez les sujets âgés, présence d'une auto-immunité aspécifique.
Tous les types histologiques de thymomes ne sont pas associés de manière identique à une myasthénie, certains n'y étant même jamais associés[20] :
- type A : 0 % ;
- type AB : 6,8 % ;
- type B1 : 40 % ;
- type B2 : 55,6 % ;
- type B3 : 10 % ;
- carcinomes thymiques : 0 %.
Maladies auto-immunes
Dans 5 à 15 % des cas selon les séries, au moins une autre maladie auto-immune est identifiée chez les myasthéniques. Il s'agit de rapports d'associations significatives, aucun lien physiopathologique n'ayant été démontré hormis l'hypothèse d'un « terrain dys-immun ». Ces pathologies sont les dysthyroïdies auto-immunes, la polyarthrite rhumatoïde, le lupus érythémateux disséminé, le syndrome de Sjögren, la polymyosite, les maladies intestinales inflammatoires chroniques, l'anémie de Biermer, l'anémie hémolytique auto-immune, l'insuffisance surrénalienne.
Examens complémentaires
Les examens complémentaires, outre ceux visant à écarter d'éventuels diagnostics alternatifs, ont pour but d'affirmer ou d'exclure le diagnostic et, en cas de confirmation diagnostique, de rechercher des pathologies associées. Ces examens sont[6],[21],[22],[23],[24],[25],[26],[27],[28],[29] :
Biologie
Seuls les anticorps anti-récepteur à l'acétylcholine et, s'ils sont absents, les anticorps anti-MuSK sont recherchés en pratique clinique. Leur spécificité est de 99 %. Leur sensibilité globale est de plus de 90 %, moins de 10 % des myasthénies étant donc définies comme « séronégatives ». Plus précisément, la sensibilité des anticorps anti-récepteur à l'acétylcholine est de 90 % pour les myasthénies généralisées et de 50 % pour les myasthénies oculaires. À la condition que la recherche d'anticorps anti-récepteur à l'acétylcholine soit négative, la sensibilité des anticorps anti-MuSK est de 40 % pour les myasthénies généralisées et marginale pour les myasthénies oculaires.
Leurs excellentes spécificités en font le meilleur test permettant d'affirmer le diagnostic en cas de forte suspicion clinique (valeur prédictive positive de seulement 1 % pour la population générale).
Depuis peu, en cas de myasthénie double négative, on peut aussi doser les anticorps LRP4[30].
Examens électrophysiologiques
Deux examens électrophysiologiques sont nécessaires :
- une étude électromyographique qui comprend :
- un électromyogramme de routine qui est généralement normal. Des activités de réinnervation peuvent cependant être observées dans les cas sévères évolués,
- une étude électromyographique avec stimulation répétitive supra-maximale d'un nerf moteur. Le test est considéré comme positif lorsque le décrément d'amplitude du potentiel d'unité motrice (PUM) est supérieur à 10 %. Ce test est aspécifique et présente une sensibilité de 79 % pour les myasthénie généralisées et de 50 % pour les myasthénies oculaires,
- une étude électromyographique « fibre unique » avec mesure d'un Jitter (variation de la latence entre la stimulation et le Potentiel d'Unité Motrice pour une fibre donnée lors d'une stimulation répétitive d'un nerf moteur) moyen sur l'étude de nombreuses fibres nécessitant un examinateur expérimenté et un matériel spécialisé. Le test est considéré comme positif lorsqu'est observée une élévation de ce jitter au-dessus des normes du laboratoire pour le muscle étudié. Ce test est non spécifique et présente une sensibilité de 98 % pour les myasthénies généralisées et de 80 % pour les myasthénies oculaires,
- une étude des vitesses de conduction nerveuses. Cet examen doit être normal pour que des anomalies à l'électromyogramme soient considérées pertinentes.
L'excellente sensibilité de l'électromyographie en fait le meilleur test permettant d'exclure le diagnostic en cas de faible suspicion clinique (valeur prédictive négative indéterminable si la proportion de malades dans le groupe étudié est supérieure à 1 %).
Test pharmacologique
La prostigmine est utilisée soit comme test thérapeutique per os, soit en IM ou surtout IV devant un symptôme clinique qui n’a pas parlé sur le plan électro physiologique ou en urgence (ptosis, diplopie, troubles phonatoires ou respiratoires).. Le test est considéré comme positif s'il entraîne une amélioration des symptômes ou des signes électrophysiologiques. Il est parfois comparé à un test avec placebo.
Biologie
Les marqueurs des pathologies auto-immunes potentiellement associées à la myasthénie seront systématiquement dosés. Cela comprend les marqueurs d'inflammation, les hormones thyroïdiennes et anticorps antithyroperoxydases, les facteurs antinucléaires, les anticorps anti-nucléaires, un hématogramme, un ionogramme, les fonctions hépatiques et rénales, la vitamine B12 et les folates.
CT-scanner thoracique
Réalisé systématiquement afin de rechercher un thymome (sensibilité de 99 %, spécificité de 95 %) et une hyperplasie thymique (sensibilité de 50 %, spécificité de 95 %).
Diagnostic différentiel
Le diagnostic différentiel est large et dépend de la présentation clinique. On peut notamment évoquer[31] :
- autres syndromes myasthéniques : myasthénie congénitale, syndrome myasthénique de Lambert-Eaton, le botulisme, certains venins de serpent, les intoxications aux organo-phosphorés ou au magnésium ;
- les myopathies ;
- les neuropathies périphériques ;
- les pathologies rhumatismales ;
- des troubles hydro-électrolytiques ;
- les endocrinopathies ;
- les anémies ;
- les ptosis acquis, congénitaux et traumatiques ;
- les strabismes décompensés ;
- les troubles fonctionnels, les troubles psychiatriques et le syndrome de fatigue chronique.
Pronostic
Le pronostic est propre à chaque patient. On peut cependant décrire quelques généralités[5],[6],[11],[12],[13],[14] :
La gravité de la myasthénie est liée à la survenue de crises myasthéniques avec détresse respiratoire et risque vital. Ce risque est maximal en début de maladie, 75 % des crises survenant dans les deux premières années d'évolution.
En l'absence de traitement, l’évolution des myasthénies généralisées peut être fatale. Le pronostic vital des formes demeurant oculaires n'est quant à lui pas engagé.
Il existe dans de très rares cas, des rémissions spontanées de la maladie dont la durée est variable.
10 à 20 % répondront peu ou pas aux traitements et développeront une impotence motrice variable. Les facteurs de mauvais pronostic et de mauvaise réponse thérapeutique sont : la présence d'un thymome, la présence d'anticorps anti-MuSK et les formes séro-négatives.
Prise en charge thérapeutique
La prise en charge de la myasthénie peut se diviser schématiquement en un traitement symptomatique, un traitement de fond et un traitement de crise de la maladie, en un traitement des pathologies associées et en des mesures générales. Les principes généraux en sont[32],[33],[12],[34],[35] :
Anti-cholinestérasiques
Inhibiteurs de l'acétylcholinestérase, enzyme dégradant l'acétylcholine, ils constituent le traitement symptomatique de première intention. Les plus utilisés sont la pyridostigmine (Mestinon) et le chlorure d’ambénonium (Mytélase)[36]. L'utilisation des autres anticholinestérasiques (telle que la néostigmine) est devenue marginale hors crise myasthénique. Leur efficacité dans les myasthénies séronégatives ou avec anticorps anti-MuSK n'est pas démontrée.
Corticoïdes et immunosuppresseurs
Les corticoïdes — tel qu'un schéma progressif de prednisone (Cortancyl) ou prednisolone (Solupred) — les immunosuppresseurs — l'azathioprine (Imurel) ayant le plus démontré son efficacité[34] — et le mycophénolate mofétil (Cellcept) constituent le traitement de fond de deuxième intention. Les corticoïdes peuvent être débutés à très faibles doses et avec réévaluation clinique rapprochée, une aggravation clinique transitoire étant courante.
Il est aujourd'hui démontré que l'adjonction d'un traitement combiné par corticoïdes et immunosuppresseurs pour les myasthénies généralisées insuffisamment contrôlées sous anticholinestérasiques améliorent l'état clinique à deux ans par rapport à l'adjonction de corticoïdes ou d'immunosuppresseurs séparément. Certains experts recommandent leur utilisation en première intention pour les formes séronégatives ou avec anticorps anti-MuSK.
Les immunosuppresseurs ne sont pas très efficaces pour les formes oculaires.
De façon marginale, des traitements biologiques tels que le rituximab (Mabthera), anticorps monoclonal chimérique ciblant la molécule CD20 exprimée spécifiquement par les lymphocytes B, ont été utilisés avec un certain succès dans des formes graves[37].
Plasmaphérèses et Immunoglobulines intraveineuses
Ces traitements n'ont que peu de place en dehors des crises myasthénique (cf. infra). Ils sont cependant exceptionnellement utilisés pour les formes les plus sévères résistantes aux autres traitements ou en cas de dégradation clinique rapide.
Thymectomie et traitements adjuvants
La découverte d'un thymome est une indication absolue de thymectomie et nécessite un suivi radiologique prolongé à la recherche de récidives néoplasiques. En cas de résection incomplète, d'infiltration des tissus avoisinant ou de découverte de métastases, un traitement adjuvant, consistant en une radiothérapie et/ou une chimiothérapie selon les cas, doit être proposé.
L’intérêt clinique de la thymectomie en l’absence de thymome est encore discuté. Elle est cependant recommandée en cas de découverte d'une hyperplasie thymique avec présence d'anticorps anti-récepteurs à l'acétylcholine chez les patients de moins de 40 ans. Une étude randomisée de 2016 retrouve par ailleurs une amélioration clinique des symptômes de la myasthénie après thymectomie, même en absence d'hyperplasie thymique[38].
Une aggravation symptomatique post-chirurgicale transitoire est possible. Dans les premiers mois suivant la thymectomie, un sevrage médicamenteux peut être tenté, si une amélioration symptomatique voire une rémission apparait.
Autres traitements
Des traitements spécifiques sont nécessaires en cas de découverte d'une pathologie auto-immune associée.
Prise en charge supportive
Toute installation rapide d'une insuffisance respiratoire, d'une dyspnée avec encombrement bronchique, de troubles de la déglutition ou d'une faiblesse généralisée objectivée par un médecin justifie un transfert médicalisé vers une institution hospitalière disposant d'une unité de soins intensifs. À l'admission, une évaluation des voies respiratoires, de la respiration, de la circulation et neurologique sera réalisée ainsi qu'une mesure de la capacité vitale (CV). Un trouble de la conscience, une capacité vitale inférieure à 25 % de la valeur théorique ou un épuisement respiratoire clinique justifient une intubation et un transfert en unité de soins intensifs pour ventilation mécanique et éventuelle stabilisation hémodynamique. Une prise en charge ordinaire avec évaluation régulière de la capacité vitale suffit dans les autres cas.
Correction des facteurs déclenchants et favorisants réversibles
Ces facteurs doivent être recherchés et corrigés dès les manœuvres vitales assurées. Ce sont principalement les causes iatrogènes (cf Contre-indications médicamenteuses), les infections, la décompensation d'autres pathologies chroniques du patient et les élévations de températures, endogènes ou exogènes.
Le cas des crises survenant lors de grossesses est particulier et la possibilité de leur survenue doit avoir été discutée préalablement avec la patiente. Les crises sévères ne répondant pas au traitement symptomatiques nécessitent une prise en charge adaptée.
Exclure un surdosage en anticholinestérasiques
Le principal diagnostic différentiel de la crise myasthénique est le surdosage en anticholinestérasiques. Il est à suspecter systématiquement en cas de prise quotidienne de plus de 480 mg de pyridostigmine ou de symptômes muscariniques : bradycardie, myosis bilatéral, hypersécrétions, débâcle fécale, etc. En cas de doute, une aggravation clinique après administration de néostigmine confirmera le diagnostic de surdosage. La prise en charge est alors purement supportive, hors administration d'atropine pour ses effets anti-muscariniques.
Traitements symptomatiques
L'administration de néostigmine constitue le traitement symptomatique de première intention. En cas d'apparition de symptômes muscariniques, on lui adjoindra de l'atropine.
En cas d'aggravation clinique sous traitement, un surdosage en anticholinestérasique doit être évoqué.
En cas de mauvaise réponse thérapeutique (échec de détubation), le traitement de seconde intention repose sur la répétition de plasmaphérèses et/ou de cures d'immunoglobulines intraveineuses.
En cas d'échec, les corticoïdes et les immunosuppresseurs constituent le traitement de troisième intention. Leurs indications doivent être cependant prudemment posées, une aggravation clinique transitoire étant courante.
Carte de myasthénique
Le ministère de la santé français a édité une carte de soins et d’urgence pour les personnes myasthéniques. Le port par le patient de cette carte mentionnant sa maladie, son traitement, ses dernières vaccinations et la liste des médicaments contre-indiqués dans la myasthénie est vivement recommandé. Ils peuvent se la procurer auprès des consultations de référence ou en contactant l’AFM-téléthon].
Hygiène de vie
Comme pour toute pathologie chronique, la myasthénie nécessite une éducation du patient à sa maladie et particulièrement aux facteurs favorisants de crises myasthéniques. Une participation à de l'exercice physique modéré, adapté en fonction de l'état du moment, doit être encouragée avec des repos fréquents[39].
Prise en charge para-médicale
Dans les formes sévères ou réfractaires aux traitements de fonds, une prise en charge sociale et par des spécialistes en kinésithérapie, en orthophonie, en ergothérapie, en revalidation ou en psychologie peut s'avérer nécessaire tout comme l'orientation du patient vers une association de malades.
Préparer les grossesses et les chirurgies
De façon générale, la grossesse et les interventions chirurgicales sont une cause importante de crises myasthéniques, qui surviennent dans 30 % des cas. Afin de minimiser les risques, certains auteurs recommandent la réalisation pré-partum et pré-chirurgicale de séances de plasmaphérèses ou de cures d’immunoglobulines ainsi que d'une évaluation comprenant des épreuves fonctionnelles respiratoires. Un accouchement doit toujours se dérouler dans une institution hospitalière disposant d'une unité de soins intensifs et d'une unité de réanimation néo-natale. Une détubation ne doit jamais être effectuée avant que la capacité du patient à y participer soit affirmée.
Durant la grossesse, les corticoïdes et les immunosuppresseurs, doivent impérativement être employés en concertation avec un gynécologue.
Notes et références
- (en) MB Walker, « Treatment of myasthenia gravis with physostigmine », Lancet, vol. 1, , p. 1200-1201 (DOI 10.1016/S0140-6736(00)94294-6)
- (en) Phillips LH, « The epidemiology of myasthenia gravis » Ann N Y Acad Sci. 2003;998:407-12
- Collège des enseignants de neurologie. Myasthénie
- (en) Carr AS, Cardwell CR, McCarron PO, McConville J, « A systematic review of population based epidemiological studies in myasthenia gravis » BMC Neurol. 2010;10:46.
- (en) Bradley et al. Neurology in clinical practice, 5e éd., Butterworth-Heinemann, 2007
- Goulon-Goeau et al. « Myasthénie et syndromes myasthéniques » EMC-Neurologie, 17-172-B-10, 2002, Elsevier, résumé en ligne
- B. Eymard « Anticorps dans la myasthénie » Revue neurologique 2009;165(2):125-205 DOI:10.1016/j.neurol.2008.11.020
- (en) Zhang B, Shen C, Bealmear B, Ragheb S, Xiong WC, Lewis RA, Lisak RP, Mei L. « Autoantibodies to agrin in myasthenia gravis patients » PLoS One, 2014;3:e91816 DOI:10.1371/journal.pone.0091816
- (en) W. Allan. « Pathogenesis of myasthenia » Up-to-Date, 2012
- (en) Howard et al. « Clinical correlations of antibodies that bind, block or modulate human acetylcholine receptors in myasthenia gravis » Ann NY Acas Sci. 1987
- (en) S.J. Bird « Clinical manifestations of myasthenia gravis » Up-to-Date, 2013
- (en) Bershad et al. « Myasthenia gravis crisis » South Med J. 2008;101(1):63-9. DOI:10.1097/SMJ.0b013e31815d4398
- Gaumond « Troubles oculaires de la myasthenie » EMC-Neurologie, 2005 DOI:10.1016/j.emcn.2005.08.002
- (en) Pelak et Quand « Ocular myasthenia » Up-to-Date, 2013
- (en) Spillane J, Higham E, Kullmann DM. « Myasthenia gravis » BMJ 2012;345:e8497
- (en) Jaretzki A, Barohn RJ, Ernstoff RM et al., « Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America », Neurology, vol. 55, no 1, , p. 16–23 (PMID 10891897, lire en ligne)
- (en) Finnis MF, Jayawant S, « Juvenile myasthenia gravis: a paediatric perspective », Autoimmune Dis, vol. 2011, , p. 404101. (PMID 22110902, PMCID PMC3206364, DOI 10.4061/2011/404101, lire en ligne [html])
- Antititin antibody in early- and late-onset myasthenia gravis. Szczudlik P, Szyluk B, Lipowska M, Ryniewicz B, Kubiszewska J, Dutkiewicz M, Gilhus NE, Kostera-Pruszczyk A.Acta Neurol Scand. 2014 (Juin). Doi : 10.1111/ane.12271. [Epub ahead of print]
- The thymus, thymoma and myasthenia gravis Fujii Y, Surg Today 2013 May;43(5):461-6. doi: 10.1007/s00595-012-0318-2
- Clinical and functional significance of WHO classification on human thymic epithelial neoplasms: a study of 146 consecutive tumors. Okumura M, Miyoshi S, Fujii Y, Takeuchi Y, Shiono H, Inoue M, Fukuhara K, Kadota Y, Tateyama H, Eimoto T, Matsuda H, Am J Surg Pathol 2001 Jan;25(1):103-10.
- (en) Mercelis et al. « Diagnostic utility of stimulated single-fiber electromyography of the orbicularis oculi muscle in patients with suspected ocular myasthenia » Muscle Nerve 2011;43(2):168-70.
- (en) Preston et al. Electromyography and neuromuscular disorders. Clinical-electrophysiologic correlations, 2e éd., Elsevier, 2005
- (en) Taraldsen Heldan. « Seropositive Myasthenia Gravis. A Nationwide Epidemiological Study » Neurology 2009
- (en) Trakas et al. « Development of a highly sensitive diagnostic assay for muscle specific tyrosine kinase (MuSK) autoantibodies in myasthenia gravis » J Neuroimmunol. 2011;240-241:79-86.
- (en) Vincent et al. « Acetylcholine receptor antibody as a diagnostic test for myasthenia gravis : results in 153 validated cases and 2967 diagnostic essays » J Neurolo Neurosurg Psych. 1985;48(12):1246-52.
- (en) Witoonpanich et al. « Electrophysiological and immunological study in myasthenia gravis: diagnostic sensitivity and correlation » Clin Neurophysiol. 2011;122(9):1873-7.
- (en) Zambelis et al. « Repetitive nerve stimulation of facial and hypothenar muscles: relative sensitivity in different myasthenia gravis subgroups » Eur Neurol. 2011;65(4):203-7.
- (en) Lindstrom et al. « Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates and diagnostic value » Neurology 1976.
- (en) Bird « Diagnosis in myasthenia gravis » Up-to-Date, 2013
- Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Zhang B1, Tzartos JS, Belimezi M, Ragheb S, Bealmear B, Lewis RA, Xiong WC, Lisak RP, Tzartos SJ, Mei L. Arch Neurol. 2012 Apr;69(4):445-51. doi: 10.1001/archneurol.2011.2393. Epub 2011 Dec 12
- (en) Bird « Differential Diognosis of myasthenia gravis » Up-to-Date, 2012
- (en) Bird « Treatment of myasthenia » Up-to-Date, 2013
- (en) Howard et al. Myasthenia Gravis. A manual for the health care provider, Myasthenia Gravis Foundation of America, 2008
- (en) Gold R, Schneider-Gold C, « Current and future standards in treatment of myasthenia gravis », Neurotherapeutics, vol. 5, no 4, , p. 535-41. (PMID 19019304, PMCID PMC4514693, DOI 10.1016/j.nurt.2008.08.011, lire en ligne [html])
- (en) Mandawat et al. « Comparative analysis of therapeutic option used for myasthenia gravis » Ann Neurol. 2010;68(6):797-805.
- « Ambénonium chlorure », sur www.vidal.fr (consulté le )
- (en) Collongues N, Casez O, Lacour A. et al. « Rituximab in refractory and non-refractory myasthenia: a retrospective multicenter study » Muscle Nerve 2012;46:687-91
- (en) Gil I. Wolfe, Henry J. Kaminski, Inmaculada B. Aban et al., « Randomized Trial of Thymectomy in Myasthenia Gravis », New England Journal of Medicine, (NEJM\/MMS), vol. 375, no 6, , p. 511-522 (ISSN 0028-4793, DOI 10.1056/nejmoa1602489, lire en ligne).
- (en) Goldenberg, W.D. and Shah, A.K., « Myasthenia Gravis », eMedicine (consulté le )
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Myasthenia gravis » (voir la liste des auteurs).
Voir aussi
Article connexe
Lien externe
 Portail de la médecine
Portail de la médecine