Prion (protéine)
Le prion est un type de protéine dont la conformation ou le repliement est anormal et capable de transmettre cette forme mal repliée à des variantes normales de la même protéine. Ces prions dits pathogènes caractérisent plusieurs maladies neurodégénératives mortelles et transmissibles chez l'homme et de nombreux autres animaux. On ignore encore ce qui cause le mauvais repliement de la protéine normale, mais la structure tridimensionnelle anormale est soupçonnée de conférer des propriétés infectieuses, en reconfigurant les molécules de protéines voisines dans la même forme. Le rôle infectieux d'une protéine contraste avec tous les autres agents infectieux connus tels que les viroïdes, les virus, les bactéries, les champignons et les parasites, qui eux contiennent tous des acides nucléiques (ADN, ARN ou les deux comme support de l’information infectieuse). Le terme « prion » dont la paternité revient à Stanley Prusiner et remonte à 1982, serait soit l’acronyme de PROteinaceous INfectious particle (particule protéique infectieuse)[1],[2] — « pro-in » étant réarrangé en « prion » pour en simplifier la prononciation[3] —, soit, selon d'autres sources, l'acronyme de PRoteinaceous Infectious ONly[4].
Pour les articles homonymes, voir prion.
| Clade | Biota |
|---|---|
| Super-domaine | Acytota |
| Domaine | Aminoacuea |
Taxons de rang inférieur
Voir le texte
On distingue les prions de mammifères qui infectent l'humain et différentes espèces animales, des prions retrouvés chez les champignons comme chez Saccharomyces cerevisiae (levure de boulanger).
Les prions de mammifères sont les agents causaux responsables des encéphalopathies spongiformes transmissibles (EST) ou maladies à prion. Parmi les EST les plus connues, on peut citer :
- chez l’humain :
- les différentes formes de la maladie de Creutzfeldt-Jakob,
- l’insomnie fatale familiale (IFF),
- le syndrome de Gerstmann-Sträussler-Scheinker (SGSS),
- le Kuru ;
- chez l’animal :
- la tremblante du mouton et de la chèvre,
- l’encéphalopathie spongiforme bovine (ESB),
- l’encéphalopathie spongiforme féline,
- l’encéphalopathie spongiforme du vison et,
- le dépérissement chronique du cervidé (CWD pour Chronic Wasting Disease).
L’ensemble de ces maladies se caractérise par une dégénérescence du système nerveux central (cerveau et moelle épinière) liée à la propagation ou multiplication de prions chez l’hôte infecté.
D'un point de vue anatomopathologique, on observe ainsi au niveau de l'encéphale la formation de vacuoles (donnant un aspect spongieux au cerveau, d'où le nom de spongiforme dans EST), une mort des neurones, une gliose (multiplication des astrocytes et de la microglie) et l'accumulation d'une protéine de l'hôte, la PrPC (abréviation de protéine prion cellulaire, l'isoforme normale), sous une conformation anormale (ou mal repliée) alors dénommée PrPSc (abréviation de protéine prion de la scrapie)[5].
Des prions atypiques, apparemment également pathogènes mais probablement sporadiques, ont aussi été observés au moment de la percée d'ESB de 2011 en Europe, au Japon, aux États-Unis et au Canada[6].
Historique
- 1732 : première description de la tremblante.
- 1917-1918 : première description de la maladie de Creutzfeldt-Jakob.
- 1936 : première description du syndrome de Gerstmann–Sträussler–Scheinker.
- 1938 : démonstration expérimentale du caractère transmissible de la tremblante. Cuillé et Chelle ont injecté à des moutons des homogénats (tissus broyés) de cerveaux de moutons morts de la tremblante. Les animaux inoculés ont à leur tour déclaré la maladie démontrant ainsi que la tremblante était due à un agent infectieux.
- 1957 : première description du kuru par Gajdusek et Zigas.
- 1959 : Hadlow note de nombreuses similitudes anatomopathologiques entre la tremblante du mouton et le Kuru. Il suggère que le kuru pourrait aussi être causé par un agent infectieux.
- 1966 : le groupe de Gajdusek démontre le caractère transmissible du Kuru (chez le chimpanzé).
- 1967 : Alper et Pattison réalisent des études d'inactivation (ou destruction) de l'agent infectieux de la tremblante par des rayonnements ionisants. Les rayonnements ionisants conduisent à des altérations et des modifications des acides nucléiques à l'origine de l'inactivation de l'agent pathogène. La dose de rayons ionisants nécessaire pour inactiver la moitié des particules infectieuses est proportionnelle à la taille du génome et donc de la taille de l'agent pathogène lui-même. Les résultats qu'ils obtiennent par cette méthode d'estimation de taille, montre que l'agent responsable de la tremblante présente des propriétés de résistance aux rayons ionisants sans commune mesure avec celles obtenues pour les virus, les bactéries et les parasites, suggérant que l'agent infectieux responsable de la tremblante est très petit (bien plus qu'un virus). Ils supposent que cet agent pourrait être dépourvu d'acide nucléique.
- 1967 : Griffith propose l'hypothèse d'une unique protéine (ou de protein only), suggérant que l'agent infectieux responsable de la tremblante pourrait être réduit à une protéine ayant adopté un repliement anormal. Cette protéine serait capable d'imprimer sa conformation anormale à la protéine de l'hôte, ce qui serait le mode de propagation de cet agent.
- 1968 : le groupe de Gajdusek montre que la maladie de Creutzfeldt-Jakob est également transmissible. Ceci permit de regrouper les EST sous cette terminologie et d'établir qu'elles étaient dues à un agent infectieux.
- 1978 : première description de la Chronic wasting disease (CWD).
- 1982 : le groupe de Prusiner réalise une étude systématique portant sur l'inactivation de l'agent de la tremblante après transmission au hamster syrien. Ils montrent qu'aucun traitement physique et chimique détruisant les acides nucléiques ne peut inactiver l'agent infectieux. Inversement tous les procédés chimiques et physiques détruisant ou détériorant les protéines entraînent une inactivation importante de l'agent infectieux. Prusiner propose de nommer prion ce nouveau type d'agent infectieux (vraisemblablement dépourvu d'acide nucléique et de nature essentiellement protéique). Ainsi, il remet en cause le paradigme médical de trois sortes d'agents infectieux (virus, microbes et parasites). La notion de prion est alors mal acceptée par la communauté médicale[7].
- 1982 : le groupe de Prusiner identifie une protéine qui copurifie avec l'agent infectieux. Ils nommèrent cette protéine PrP, abréviation pour l'anglais : Protease Resistant Protein (protéine résistante aux protéases), car elle présentait la particularité d'être partiellement résistante à la digestion par les protéases (enzymes qui coupent les protéines en petits peptides).
- 1991 : le groupe de Prusiner établit que la PrP n'est pas une protéine codée par le génome de l'agent infectieux mais par le génome de l'hôte. Ces résultats montrent que chez les individus infectés la PrP normale ou PrPC, exprimée par l'hôte, est convertie (ou subit un repliement) en PrP anormale ou PrPSc.
- 1986 : première description de l'ESB.
- 1986 : première description de l'IFF (Insomnie fatale familiale).
- 1993 : développement des premières souris avec le gène Prnp, codant la protéine PrP, invalidé. Ces souris qui n'expriment plus la PrPC ne peuvent plus contracter une maladie à prion après infection expérimentale.
- 1996 : première description du « variant de la maladie de Creutzfeldt-Jakob ».
- 2016 : premier cas mondial de maladie débilitante chronique (MDC) confirmé chez un renne femelle le [8] (en Norvège), qui est aussi le 1er cas de MDC détecté en Europe chez un cervidé sauvage. Peu après sont signalés deux cas chez l'élan (en Norvège également, près de la frontière suédoise)[9].
Protéines normales et pathologiques
Le prion pathologique ou protéine PrPSc est une forme spéciale de la protéine normale PrPC qui est présente à l’état naturel et est impliquée dans le fonctionnement normal de la cellule notamment au niveau des microdomaines Raft de la membrane plasmique[10]. Les fonctions de PrPC ne sont pas encore intégralement connues mais semblent essentielles. En effet, la protéine PrPC était présente avant la spéciation des mammifères, ce qui signifie que tous les mammifères (et donc l'humain) sont susceptibles de développer des maladies à prions. La protéine PrPC est impliquée dans le développement du système nerveux chez l'embryon. Chez l'adulte, elle est exprimée essentiellement dans le cerveau et la moelle épinière (neurones et glie). Elle est impliquée dans les processus de différenciation et d’adhésion des cellules entre elles[10]. Elle aurait aussi un rôle protecteur antioxydant et vis-à-vis de la mort cellulaire programmée ou apoptose[10]. Cette protéine aurait également un rôle dans le repliement d’autres protéines.
Selon l'équipe du Dr Scott (), la protéine normale, étudiée chez le rat, présente des accumulations particulières à l'intérieur des cellules du pancréas spécialisées dans la production d'insuline, et les rats prédisposés au diabète présentent 3 fois plus de cellules productrices d'insuline avec des amas de protéines PrPC. Le taux de PrPC dans le pancréas d'un rat normal change fortement dans les un à trois jours suivant l'administration de concentrations élevées de sucre via le sang. La protéine PrPC pourrait être impliquée dans le diabète de type 1 ou juvénile, maladies caractérisées par une attaque par le système immunitaire des cellules produisant l'insuline (dans le pancréas)[11].
Le prion pathologique est une protéine PrPC repliée différemment, notée PrPSc. La PrPSc résulte d’une modification de la structure tridimensionnelle de PrPC. Elle provoque les maladies à prions (maladie de la vache folle, ou encéphalopathie spongiforme bovine, maladie de Creutzfeldt-Jakob, tremblante du mouton, Chronical Wasting Disease ou maladie du dépérissement chronique des cervidés). Lors de l'infection, l'agent prion, agent pathogène responsable de l'infection, pénètre le neurone, où pour des raisons et par un mécanisme encore mal compris il se multiplie, en dépliant/repliant les protéines PrPC en protéines PrPSc, forme qui n'est plus dégradée par protéolyse et qui, par accumulation dans la cellule, finit par la tuer et former des plaques de dépôts dans le cerveau.
Dans toutes ces maladies, aucun acide nucléique (ADN/ARN) n’a pu être spécifiquement associé à l’infectiosité, comme a pu l'être la protéine PrPSc. On parle d'agent transmissible non conventionnel (ATNC).
Les maladies à prions sont transmissibles d’un individu à l'autre et dans une certaine mesure d'une espèce à l’autre.
Deux formes atypiques de prions pathogènes nommées BSE-H et low-type BSE-L, aux symptômes proches chez l'animal de laboratoire de ceux de l'ESB, mais différents de ceux qui causent l'ESB, probablement sporadiques ont été récemment découvertes[6]. Leur origine bovine confirme que l'agent de l'ESB peut effectivement être lui-même un prion bovin muté.
Écoépidémiologie du prion
Au moins un prion pathogène (celui qui cause la maladie débilitante chronique des cervidés) affecte la faune sauvage en liberté et circule dans l'environnement, en Amérique du Nord, et depuis peu en Europe du Nord[8].
On cherche encore à mieux comprendre l'occurrence de ce prion[12] et son « écologie »[13] ainsi que celles d'autres prions pathogènes (provenant d'élevages bovins et ovins) et la manière dont ils circulent dans l'environnement[14],[15] où l'on sait qu'ils peuvent persister durant plus de 15 ans[16], notamment dans le sol où ils restent infectieux[17],[18].
Avec l'amélioration des seuils de sensibilité des outils de dosages, l'évaluation des charges environnementales de prions dans le sol et l'eau a commencé à se préciser dans les années 2000. TA Nichols et al. ont ainsi en 2009 recherché et détecté le PrPCWD dans de l'eau prélevée dans une zone d'endémie de la maladie débilitante chronique (CWD), durant la fonte des neiges[19]. Les tests (de type bioessai) ont indiqué des taux de PrPCWD inférieurs aux seuils considérés comme infectieux, mais confirmant la présence à bas bruit (« très faibles concentrations ») de PrPCWD dans l'environnement, la persistance et l'accumulation de prions dans l'environnement[19], posant par exemple la question d'éventuelles accumulation dans le sédiment ou une éventuelle bioconcentration par des organismes aquatiques filtreurs (moules d'eau douce, éponges d'eau douce, larves d'invertébrés...).
Rôles écoépidémiologiques des végétaux
En 2014, Rasmussen et al. se demandent si les plantes pourraient contribuer à la dispersion de maladies à prion[20] ; après avoir exposé des plants de blé intacts à des prions infectieux (PrPTSE) durant 24 h dans trois études répétées avec PrPTSE, le prion a été retrouvé lié aux racines, mais n'a pas pu être détecté dans la tige ni les feuilles par les méthodes employées. Les auteurs ont conclu que dans le cas du blé, si des prions sont transportés des racines aux tiges, ils le sont à des niveaux inférieurs aux seuils détectables par le Western blot et les kits de diagnostic IDEXX ou Bio-Rad.
Par contre une étude faite aux États-Unis (où un grand nombre de cervidés porteurs ou potentiellement porteurs de prions pathogènes ont été tués et enterrés, pour lutter contre l'extension de la CWD, ou "cachexie chronique"), portée par le Centre des sciences de la santé de l'Université du Texas (Houston) a en effet montré en 2015 que les plantes sont aussi vectrices de prions, et potentiellement de deux manières :
- Des prions peuvent avoir été déposés sur la partie aérienne de la plante, via des excrétats (mucus, salive...), excréments ou urines d'animaux malades. En laboratoire, des feuilles sur lesquelles on a pulvérisé une préparation contenant des prions pathogènes les ont conservés durant plusieurs semaines à l'intérieur de la plante vivante ; les animaux qui consomment ces plantes peuvent alors se contaminer ; en 2016 Ortega publie une étude basée sur la PMCA (Protein misfolding cyclic amplification) comme moyen de détecter la présence éventuelle de prions pathogènes dans diverses graminées et plantes naturellement exposées aux prions dans les zones où la CWD est devenue endémique. Le PrPRES est effectivement retrouvé en surface de plusieurs plantes du Parc national de Rocky Mountain ; et les souris inoculées avec ces échantillons développent la maladie[21] ;
- D'autre part le prion pathogène peut aussi être libéré dans le sol à partir de cadavres en décomposition, puis être possiblement absorbé via les racines : en effet des hamsters alimentés avec des plantes croissant sur un sol dans lequel était enterré un cerf mort de CWD (équivalent de la vache folle chez le cerf) sont tombés malades, ce qui évoque une migration du prion du sol vers la plante[22],[23] ; On sait depuis les années 1960 que les plantes peuvent capter via leurs racines certaines protéines[24],[25], dont pour en faire une source d'azote dans certains cas[26] (elles peuvent aussi absorber des enzymes[25] et des microbes[20]). De petites quantités de PrPSc contenues dans un homogénat de cerveau dilué ou dans des matières excrétoires (urine et fèces) peuvent se lier aux racines ainsi qu'aux feuilles[27].
Les auteurs ont observé des interactions prion-plantes avec des prions d'origines diverses (y compris le prion de la CWD)[27]. Ils signalent que des hamsters de type sauvage ont été expérimentalement effectivement infectés en ingérant des plantes contaminées par des prions (plantes exposées à des prions pathogènes puis lavées 5 fois complètement et séchées avant d'être données comme aliment aux hamsters)[27].
Ils concluent que les plantes pourraient donc jouer un rôle dans la transmission horizontale de la maladie[27].
Incinération
Les animaux atteints de maladies à prions doivent suivre un cursus d'incinération spécifique[28]. C'est ainsi que les animaux (humains inclus) non diagnostiqués ou mal diagnostiqués peuvent contribuer à la propagation de maladies, les prions pouvant résister aux températures de processus classique. En associant les rejets des funérariums et les rôles écoépidémiologiques des végétaux, les pluies contribueraient à la diffusion des prions dans l'alimentation[réf. nécessaire].
Maladies
Troubles dus à sa présence
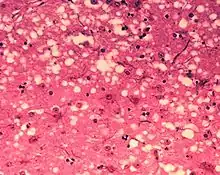
Les maladies à prions provoquent une dégénérescence du système nerveux central qui est toujours fatale :
- le rôle de prions est établi dans certaines affections animales telles que l'encéphalopathie spongiforme bovine (ESB ou maladie de la vache folle), la tremblante du mouton et de la chèvre, et la maladie du dépérissement chronique des cervidés par Stanley Prusiner ;
- chez l’humain, un prion est responsable de la maladie de Creutzfeldt-Jakob qui se caractérise par une démence précoce aboutissant au décès. La forme commune est sporadique (80 % des cas), atteignant le plus souvent le sujet âgé. Elle peut être rarement familiale, avec dans ce cas une implication du gène de la protéine prion. Elle peut être également transmise par inoculation de tissus contaminés (extraits d’hypophyse auparavant employés dans le traitement par l’hormone de croissance, greffes de cornée et de dure-mère, électrodes contaminées).
En mars 1996, est apparue une forme clinique chez le sujet jeune (< 30 ans), appelé nouveau variant de la maladie de Creutzfeld-Jakob, dont le lien avec l’ESB a été prouvé ensuite. La transmission serait due probablement à l’ingestion de viande bovine contaminée par l’ESB. Le prion est également la cause d’autres maladies humaines : le kuru aujourd’hui disparu (touchant des tribus Foré de Nouvelle-Guinée qui avaient la particularité culturelle de manger le cerveau de morts lors de rites anthropophages mortuaires et qui a été la 1re encéphalopathie spongiforme humaine dont la transmissibilité au singe a été démontrée), la maladie de Gertsmann-Sträussler-Scheinker et l’insomnie fatale familiale.
Il existe d’autres maladies neurologiques comportant des accumulations de protéines anormales, telles la maladie d'Alzheimer et la maladie de Parkinson. La responsabilité d’un prion n’a toutefois pas été démontrée dans ces cas, bien qu’il puisse coexister.
Troubles dus à l’absence de PrPC
Les données disponibles proviennent d’expérimentation de transgenèse sur des souris/hamsters à qui on a retiré le gène de la protéine PrPC et qui donc ne possèdent plus cette protéine, ou dont on peut stopper à volonté la production de protéine PrPC. Ces travaux permettent d’élucider peu à peu les fonctions de la protéine. Certaines souris dépourvues de protéine par knock-out du gène prnp codant cette protéine, sont viables et fertiles, sans phénotype apparent. D'autres développent une mort neuronale massive au niveau du cervelet. Cette mort est due à une autre protéine, paralogue à la protéine saine PrPC, appelée Doppel (Dpl).
Ce sont plus des modèles expérimentaux que de véritables prions puisqu’il manque dans ces cas la notion d'«infection». Les « PrPC » de levure ne forment pas des protéines prion comme chez les animaux, mais sont en réalité des protéines (souvent de choc thermique) qui en miment le comportement : dans certaines conditions de stress, elles changent de conformation et s'accumulent, perturbant le fonctionnement cellulaire de la levure.
Mécanismes
Quand la machinerie et les composants nécessaires (ARN-polymérase, ribosome, etc.) sont présents, il est possible de fabriquer des protéines à partir de l’ADN conformément au programme qu’il contient. Toutefois, à composition identique, une protéine peut posséder plus d’une façon de se replier, soit des conformations différentes.
On a constaté que la protéine prion anormale favorise un type de repliement anormal. Or la fonctionnalité de la protéine dépend de son bon ou mauvais repliement.
Le plus puissant ordinateur du monde (en 2004), Blue gene, a été commandé par le Laboratoire national de Lawrence Livermore pour étudier de façon systématique, par simulation, les repliements de protéines en présence et en l’absence de prions.
La levure de bière pourrait être un modèle expérimental intéressant : certaines de ses protéines ont des propriétés de « contagion de forme » qui évoquent celles des prions, même si l’assimilation à ces dernières est discutée.
Diagnostic
Le diagnostic d'une maladie à prions est fait sur des prélèvements de tissus neurologiques obtenus post-mortem. Chez l'animal, le mouton ou la vache, la technique utilisée en routine est basée sur la détection de la protéine prion pathologique par technique immunologique (ELISA et/ou Western-Blot). Les techniques biochimiques sont préférées aux techniques immunohistochimiques car elles permettent de réaliser des analyses en série sur des milliers de bêtes. Ils sont réalisés dans les laboratoires vétérinaires départementaux. Pour la vache folle, tout résultat positif est vérifié dans le laboratoire de référence de la vache folle de l'ANSES (ex AFSSA) à Lyon. Chez l'humain, on utilise trois types de techniques permettant de faire le diagnostic de maladie à prions :
- La mise en évidence d'une triade de signes neuropathologiques (perte neuronale, astrogliose, vacuoles spongiformes) ;
- La mise en évidence de dépôts de protéine prion par technique immunoshistochimique ;
- La mise en évidence de la protéine prion pathologique après traitement à la protéinase K par technique Western-Blot.
Les deux premières techniques sont généralement faites en France dans douze services d'anatomocytopathologie membres d'un réseau focalisé sur les maladies à prions humaines ; en France, les techniques biochimiques ne sont réalisées que dans deux laboratoires hospitaliers équipés d'installations P3 dédiées uniquement aux agents transmissibles non conventionnels (Groupement hospitalier Est, HCL, Lyon et La Pitié-Salpétrière, APHP, Paris)[29].
De manière beaucoup plus rare, le prion peut être détecté à partir de tissu cérébral obtenu par biopsie. Ce geste ne doit être réservé que dans des cas où un diagnostic alternatif curable (encéphalite virale....) est évoqué. Enfin, lors d'une suspicion d'une maladie de Creutzfeldt-Jakob liée à la vache folle (v-MCJ), la recherche de la protéine prion pathologique peut être réalisée sur du tissu d'amygdale obtenue par amygdalectomie. Ce geste invasif ne doit être réalisé qu'avec des arguments solides pour la suspicion de v-MCJ.
Malheureusement, actuellement, du fait d'un manque de sensibilité, la recherche des prions ne se fait pas dans les liquides biologiques classiques (urines, sang ou liquide céphalo-rachidien).
En gagnant en sensibilité (détection d’un faible nombre de particules), on espère pouvoir faire, dans l’avenir, un diagnostic par une simple prise de sang sur un sujet vivant.
Recherche
Le 27 juillet 2021, l'Anses, le CEA, le CNRS, l'INRAE et l'Inserm, en lien avec les ministères de l'enseignement de la recherche et de l'innovation et de l'agriculture et de l'alimentation, ont décidé de suspendre pour trois mois l'ensemble de leurs recherches et expérimentations sur les maladies des prions. Cette décision vient à la suite de la découvert d'un cas de la maladie de Creutzfeldt-Jakob contracté par une agent à la retraite ayant travaillé dans un laboratoire sur les prions[30] Ce moratoire a été prolongé jusqu'à fin 2021 et une mission d'inspection a été diligentée[31].
Le décès de l'agent à la retraite en novembre 2021 fait suite à celui de Émilie Jaumain le qui avait contracté la maladie en 2010 lors d'un accident de manipulation[31]. Un rapport d'inspection avait été réalisé, en , sur la sécurité dans les laboratoires de recherches à la suite de l'évènement[32].
Traitements
Préventif
Il repose sur :
- la détection et l’élimination des animaux porteurs ;
- la détection des sujets à risque devant conduire à des précautions accrues lorsqu’ils nécessitent une exploration.
Un vaccin est difficile à trouver du fait de la présence de la protéine normale dans l'organisme. Des chercheurs helvétiques ont donc modifié les gènes des souris pour que leurs lymphocytes B fabriquent des anticorps qui sauront différencier un PrPSc d'un PrPC normal. Néanmoins, il n’existe à ce jour pas de vaccin, ni de sérum ayant démontré une efficacité.
Curatif
Les principaux obstacles à un traitement efficace sont qu’il s’agit de maladies de l’encéphale, séparé de la circulation sanguine par une barrière hémato-encéphalique empêchant le passage de la plupart des molécules et que le système immunitaire ne reconnaît pas ce type d'agents infectieux.
Traitements potentiels
Les progrès de la modélisation informatique ont permis aux scientifiques d'identifier des composés qui peuvent servir de traitement pour les maladies à prions. Par exemple, des chercheurs ont découvert une protéine chaperone capable de se lier à la protéine PrPC et de stabiliser cette conformation, ce qui réduit la quantité de protéine prion cytosolique (PrPSc) nuisible[33].
Éradication
Le prion est une protéine solide (plutôt de petite de taille : 30 kDa), détruite essentiellement par les hautes températures (autoclave à 134 °C pendant 18 minutes[34]) à une pression de 3 bars. C'est pourquoi il est important de respecter scrupuleusement les 134 °C pendant 18 minutes. Il existe également des méthodes chimiques telles que l’eau de Javel fraîchement diluée à 6° chlorométrique et la soude utilisées à température ambiante pendant 1 heure ou les produits qui répondent au protocole standard prion (voir le site ANSM). Ne possédant pas de métabolisme, il n’est guère vulnérable aux irradiations utilisées habituellement dans un but de stérilisation. Cependant, aucune de ces méthodes n’offre une garantie absolue ; l’efficacité maximale est obtenue en associant un traitement chimique au traitement thermique. Les déchets inactivés par ces méthodes doivent ensuite être incinérés dans un centre agréé.
En 2004, l’Institut de génétique humaine (IGH), à Montpellier, a déposé un brevet pour leur découverte de la dégradation par l’action combinée du cuivre et d’un agent oxydant comme l’eau oxygénée[35].
Divers
La recherche sur le prion a fait l’objet de deux prix Nobel de physiologie ou médecine :
- D. Carleton Gajdusek en 1976 pour ses travaux sur le kuru ;
- Stanley B. Prusiner en 1997 pour sa théorie sur le prion, protéine infectieuse.
Notes et références
- (en) Alison Abbott, « The red-hot debate about transmissible Alzheimer's », Nature, (DOI 10.1038/531294a, lire en ligne)
- (en) SP Prusiner, « Novel proteinaceous infectious particles cause scrapie », Science, (ISSN 0036-8075, lire en ligne)
- Jill-Patrice Cassuto, De la maladie de la vache folle à celle de Creutzfeld-Jakob, Odile Jacob, , p. 69.
- « DÉCOUVERTE DU PRION », sur universalis.fr,
- Jack J. Pasternak, Génétique moléculaire humaine : Une introduction aux mécanismes des maladies héréditaires, De Boeck Supérieur, , p. 301.
- Torres J-M, Andréoletti O, Lacroux C, Prieto I, Lorenzo P, Larska M, et al. Classical bovine spongiform encephalopathy by transmission of H-type prion in homologous prion protein context. Emerg Infect Dis [serial on the Internet]. Sept.2011 (Article intégral, en anglais)
- L'histoire du prion sur infodoc.inserm.fr
- Institut vétérinaire norvégien (Norwegian Veterinary Institute ou NVI), 2016, The first detection of Chronic Wasting Disease (CWD) in Europe ; CWD-Info, 05 avril 2016)
- Becker Rachel (2016) Deadly animal prion disease appears in Europe ; How brain disorder related to mad-cow disease spread to Norway is a mystery ; Nature , 18 avril 2016 doi: 10.1038 / nature.2016.19759
- (en) Roles of the cellular prion protein in the regulation of cell-cell junctions and barrier function Petit SCV, et al., Tissue Barriers (open access), 2013.
- Institut de recherche sur en santé d'Ottawa
- Gough & Maddison (2010) Prion transmission: prion excretion and occurrence in the environment | Prion, 4 (2010), pp. 275-282
- Zabel M & Ortega A (2017). The Ecology of Prions. Microbiology and Molecular Biology Reviews, 81(3), e00001-17.
- Bartelt-Hunt and Bartz, (2013) S.L. Bartelt-Hunt, J.C. Bartz Behavior of prions in the environment: implications for prion biology | PLoS Pathog., 9 (2013), p. e1003113
- Mathiason et al., (2009) Infectious prions in pre-clinical deer and transmission of chronic wasting disease solely by environmental exposure|PLoS ONE, 4 (2009), p. e5916
- Georgsson et al., (2006) Infectious agent of sheep scrapie may persist in the environment for at least 16 years |J. Gen. Virol., 87 (2006), pp. 3737-3740
- Johnson et al. (2006) Prions adhere to soil minerals and remain infectious|PLoS Pathog., 2 (2006), p. e32
- Seidel et al. (2007) Scrapie Agent (Strain 263K) can transmit disease via the oral route after persistence in soil over years| PLoS ONE, 2 (2007), p. e435
- Nichols, T. A., Pulford, B., Wyckoff, A. C., Meyerett, C., Michel, B., Gertig, K., ... & Zabel, M. D. (2009). Detection of protease-resistant cervid prion protein in water from a CWD-endemic area. Prion, 3(3), 171-183.
- Rasmussen, J., Gilroyed, B.H., Reuter, T., Dudas, S., Neumann, N.F., Balachandran,A., Kav, N.N., Graham, C., Czub, S., and McAllister, T.A. (2014).Can plants serve as a vector for prions causing chronic wasting disease ? Prion 8, 136–142.
- Ortega A.E (2016). The role of plants as an environmental reservoir of Chronic Wasting Disease prions (Doctoral dissertation, Colorado State University).
- Coockson Beecher, « Surprising’ Discovery Made About Chronic Wasting Disease », Food Safety News, (lire en ligne, consulté le )
- Sandra Pritzkow, Rodrigo Morales, Fabio Moda et al., « Grass Plants Bind, Retain, Uptake, and Transport Infectious Prions », Cell, vol. 11, no 8, , p. 1168–1175 (PMID 25981035, PMCID 4449294, DOI 10.1016/j.celrep.2015.04.036)
- Jensen, W.A., and McLaren, A.D. (1960). Uptake of proteins by plant cells—the possible occurrence of pinocytosis in plants. Exp. Cell Res. 19, 414–417.
- McLaren, A.D., Jensen, W.A., and Jacobson, L. (1960). Absorption of Enzymes and Other Proteins by Barley Roots. Plant Physiol. 35, 549–556.
- Paungfoo-Lonhienne, C., Lonhienne, T.G., Rentsch, D., Robinson, N., Christie, M., Webb, R.I., Gamage, H.K., Carroll, B.J., Schenk, P.M., and Schmidt, S. (2008). Plants can use protein as a nitrogen source without assistance from other organisms. Proc. Natl. Acad. Sci. USA 105, 4524–4529.
- Sandra Pritzkow , Rodrigo Morales , Fabio Moda , Uffaf Khan , Glenn C. Telling , Edward Hoover , Claudio Soto (2015) Grass Plants Bind, Retain, Uptake, and Transport Infectious Prions ; mis en ligne le 14 mai 2015 | Open Access CC-BY-SA 4.0 |DOI: https://dx.doi.org/10.1016/j.celrep.2015.04.036 |résumé
- « Risque de transmission des prions lors de la prise en charge d’un patient suspect de maladie de Creutzfeldt-Jakob sporadique », quotidient, (lire en ligne [PDF])
- http://www.invs.sante.fr/Dossiers-thematiques/Maladies-infectieuses/Maladies-a-declaration-obligatoire/Maladie-de-Creutzfeldt-Jakob/Reseau-national-de-surveillance-des-maladies-de-Creutzfeldt-Jakob-et-maladies-apparentees
- « Les laboratoires publics français suspendent leurs travaux sur les prions en raison d’une suspicion de cas de maladie de Creutzfeldt-Jakob », Le Monde.fr, (lire en ligne, consulté le )
- « Second décès en France dans un laboratoire travaillant sur les prions », Le Monde.fr, (lire en ligne, consulté le )
- « La sécurité dans les laboratoires de recherche sur les prions infectieux », sur agriculture.gouv.fr (consulté le )
- (en) « Hot spots in prion protein for pathogenic conversion », (consulté le )
- Association française de stérilisation
- « Une nouvelle arme contre les prions » (consulté le ).
Voir aussi
Bibliographie
- Corinne Ida Lasmézas, Qu'est-ce qu'un prion ?, Le Pommier, collection : Les Petites Pommes du Savoir n°65, (ISBN 2746502232)
- Stanley B. Prusiner, La Mémoire et la Folie : La découverte des prions - Un nouveau paradigme biologique. Odile Jacob Sciences 2015 (ISBN 978-2-7381-3156-0)
Articles connexes
- Daniel Carleton Gajdusek
- Stanley B. Prusiner
- HSP90
- Fungal prion (en)
Liens externes
- Liautard JP, Alvarez-Martinez MT, Féraudet C, Torrent J. : « La protéine prion : structure, dynamique et conversion in vitro », in: M/S médecine/sciences, 2002,18, 1, 62-69, Texte intégral.
 Portail de la biochimie
Portail de la biochimie  Portail des neurosciences
Portail des neurosciences  Portail de la médecine
Portail de la médecine  Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire