Réaction de Prins
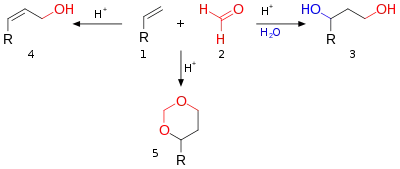
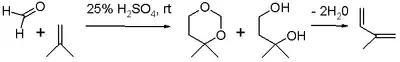
La réaction de Prins est une réaction organique consistant en une addition électrophile d'un aldéhyde ou d'une cétone à un alcène ou un alcyne, suivie de la capture d'un nucléophile ou de l'élimination d'un ion H+. Le résultat de la réaction dépend des conditions de la réaction. Avec de l'eau et un acide protique tel que l'acide sulfurique comme milieu réactionnel et du formaldéhyde, le produit obtenu de la réaction est un 1,3-diol. En l'absence d'eau, l'intermédiaire cationique perd un proton pour donner un alcool allylique. Avec un excès de formaldéhyde et une faible température de réaction, le produit de réaction est un dioxane. Lorsque l'eau est remplacée par de l'acide acétique, les esters correspondants sont formés.
Histoire
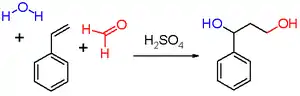
Les premiers réactifs utilisés par le chimiste néerlandais Hendrik Jacobus Prins (de) dans sa publication de 1919 étaient le styrène (schéma 2), le pinène, le camphène, l'eugénol, l'isosafrole et l'anéthole.
Hendrik Jacobus Prins a découvert deux nouvelles réactions organiques au cours de sa recherche doctorale entre 1911-1912. La première est l'addition d'un composé polyhalogène aux oléfines et la seconde réaction est l'addition catalysée par un acide d'aldéhydes à des composés oléfiniques. Les premières études sur la réaction de Prins sont de nature exploratoire et n'ont pas attiré beaucoup d'attention jusqu'en 1937. Le développement du craquage du pétrole en 1937 a augmenté la production d'hydrocarbures insaturés. En conséquence, la disponibilité commerciale d'une oléfine inférieure couplée à un aldéhyde produit à partir de l'oxydation d'une paraffine à bas point d'ébullition a augmenté la curiosité d'étudier la condensation oléfine-aldéhyde. Plus tard, la réaction de Prins est apparue comme une puissante technique de formation de liaisons CO et CC dans la synthèse de diverses molécules en synthèse organique[1].
La réaction a été étudiée dans le cadre d'une recherche en 1937 visant à utiliser des dioléfines dans le caoutchouc synthétique.
Mécanisme de réaction
Le mécanisme de réaction pour cette réaction est décrit dans le schéma 5. Le réactif carbonyle (2) est protoné par un acide protique et pour l'ion oxonium 3 résultant, deux structures de résonance qui peuvent être dessinées. Cet électrophile s'engage dans une addition électrophile avec l'alcène à l'intermédiaire carbocationique 4.
La quantité exacte de charge positive présente sur l'atome de carbone secondaire dans cet intermédiaire doit être déterminée pour chaque série de réactions.
Il existe des preuves de la participation des groupes voisins de l'oxygène hydroxyle ou de son atome de carbone voisin. Lorsque la réaction globale présente un degré élevé de concertation, la charge accumulée sera modeste.
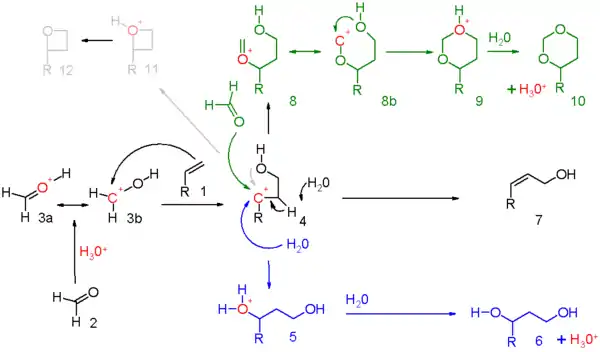
Les trois modes de réaction ouverts à cet intermédiaire oxo-carbénium sont :
- en bleu : la capture du carbocation par l'eau ou tout nucléophile approprié via 5 au 1,3-adduit 6 ;
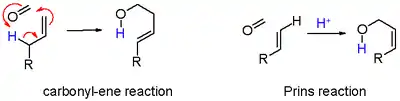
- en noir : l'abstraction de protons dans une réaction d'élimination d'un composé insaturé 7. Lorsque l'alcène porte un groupe méthylène, l'élimination et l'addition peuvent être concertées avec le transfert d'un proton allyle au groupe carbonyle qui en fait est une réaction ène dans le schéma 6 ;
- en vert : capture du carbocation par un réactif carbonyle supplémentaire. Dans ce mode, la charge positive est dispersée sur l'oxygène et le carbone dans les structures de résonance 8a et 8b. La fermeture de l'anneau conduit par l'intermédiaire 9 au dioxane 10. Un exemple est la conversion du styrène en 4-phényl-m-dioxane[2] ;
- en gris : uniquement dans des réactions spécifiques et lorsque le carbocation est très stable, la réaction prend un raccourci vers l'oxétane 12. La réaction photochimique de Paternò-Büchi entre les alcènes et les aldéhydes en oxétanes est plus simple.
Variations
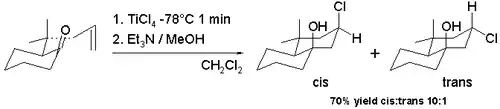
De nombreuses variantes de la réaction de Prins existent parce qu'elle se prête facilement aux réactions de cyclisation et parce qu'il est possible de capturer l'ion oxo-carbénium avec un large éventail de nucléophiles. La réaction halo-Prins est une de ces modifications en remplaçant des acides protiques et de l'eau par des acides de Lewis tels que le chlorure stannique et le tribromure de bore. L'halogène est maintenant le nucléophile se recombinant avec le carbocation. La cyclisation de certaines pulegones allyliques du schéma 7 avec du tétrachlorure de titane dans le dichlorométhane à −78 °C donne accès au squelette de la décaline avec le groupe hydroxyle et le groupe chlore majoritairement en configuration cis (91 % cis)[3]. Cette diastéréosélectivité cis observée est due à la formation intermédiaire d'un alcoxyde de trichlorotitane permettant un apport aisé de chlore à l'ion carbocation depuis la même face. L'isomère trans est préféré (98 % de cis) lorsque l'on passe à une réaction de tétrachlorure d'étain à température ambiante.
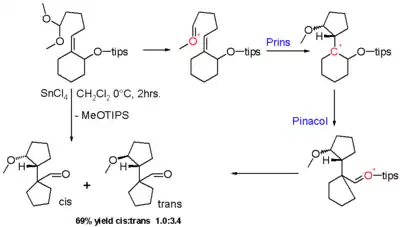
La réaction Prins-pinacol est une réaction en cascade d'une réaction de Prins et d'un réarrangement du pinacol. Le groupe carbonyle dans le réactif dans le schéma 8[4] est masqué comme un diméthyl acétal et le groupe hydroxyle est masqué sous la forme d'un éther de triisopropylsilyle (TIPS). Avec le chlorure stannique acide de Lewis, l'ion oxonium est activé et le réarrangement du pinacol de l'intermédiaire Prins résultant entraîne une contraction du cycle et un renvoi de la charge positive à l'éther TIPS qui forme finalement un groupe aldéhyde dans le produit final sous forme d'un mélange d'isomères cis et trans à diastéréosélectivité modeste.
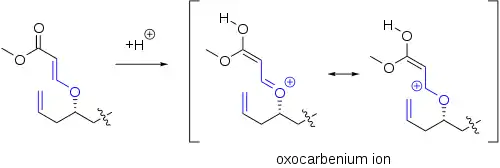
L'intermédiaire oxo-carbénium clé peut être formé par d'autres voies que la simple protonation d'un carbonyle. Dans une étape clé de la synthèse de l'exiguolide, ce dernier a été formé par protonation d'un ester vinylique[5] :

Références
- Vijaykumar S. Marakatti, « Design of solid acid catalysts for prins reaction and toluene methylation », INFLIBNET, (lire en ligne)
- R. L. Shriner et Philip R. Ruby, 4-Phenyl-m-dioxane, Organic Syntheses, coll. vol. 4, p. 786 (1963) ; vol. 33, p. 72 (1953).
- R. Brandon Miles, Chad E. Davis et Robert M. Coates, « Syn- and Anti-Selective Prins Cyclizations of δ,ε-Unsaturated Ketones to 1,3-Halohydrins with Lewis Acids », The Journal of Organic Chemistry, vol. 71, no 4, , p. 1493–1501 (PMID 16468798, DOI 10.1021/jo052142n)
- Larry E. Overman et Emile J. Velthuisen, « Scope and Facial Selectivity of the Prins-Pinacol Synthesis of Attached Rings », The Journal of Organic Chemistry, vol. 71, no 4, , p. 1581–1587 (PMID 16468809, DOI 10.1021/jo0522862)
- Min Sang Kwon, Sang Kook Woo, Seong Wook Na et Eun Lee, « Total Synthesis of (+)-Exiguolide », Angewandte Chemie International Edition, vol. 47, no 9, , p. 1733–1735 (PMID 18214872, DOI 10.1002/anie.200705018)
Voir aussi
- Hétéropolyacide
Liens externes
- Réaction de Prins dans la synthèse totale d'alcaloïdes, sur chemistry.msu.edu
- Réaction de Prins, sur organic-chemistry.org
 Portail de la chimie
Portail de la chimie