Sclérose latérale amyotrophique
La sclérose latérale amyotrophique ou SLA, également appelée dans le monde francophone maladie de Charcot (ou maladie de Lou Gehrig en Amérique du Nord), est une maladie neurodégénérative des motoneurones de l'adulte. Elle est caractérisée par une dégénérescence progressive des motoneurones du cortex cérébral avec destruction consécutive du faisceau pyramidal (atteinte du premier motoneurone) et de ceux de la corne antérieure de la moelle épinière avec destruction des unités motrices associées (atteinte du deuxième motoneurone). Elle provoque une paralysie progressive de l'ensemble de la musculature squelettique des membres, du tronc (y compris les muscles respiratoires) et de l'extrémité céphalique.
Ne pas confondre avec la maladie de Charcot-Marie-Tooth.
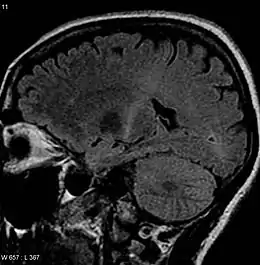
(coupe sagittale en pondération T2 FLAIR).
| Médicament | Riluzole, Édaravone, riluzole et Édaravone |
|---|---|
| Spécialité | Neurologie |
| CISP-2 | N99 |
|---|---|
| CIM-10 | G12.2 |
| CIM-9 | 335.20 |
| OMIM | 105400 |
| DiseasesDB | 29148 |
| MedlinePlus | 000688 |
| eMedicine |
1170097 emerg/24pmr/10 |
| MeSH | D000690 |
| Patient UK | Motor-Neurone-Disease-(MND) |
![]() Mise en garde médicale
Mise en garde médicale
Les causes de la SLA sont considérées comme étant multifactorielles. Elle touche les deux sexes et son incidence augmente avec l'âge à partir de 40 ans.
Aux États-Unis et au Canada, elle est également nommée « maladie de Lou Gehrig », du nom d'un joueur de baseball renommé, mort de cette maladie en 1941[1]. Également appelée « maladie de Charcot », elle ne doit pas être confondue avec une maladie de nom et de symptômes voisins, mais d'évolution moins grave, la maladie de Charcot-Marie-Tooth.
Description
Formes cliniques
Cette maladie comprend deux principales formes cliniques :
- une forme dite « spinale », débutant à l'extrémité (ou distalité) des membres (les doigts)[2] ;
- une forme dite « bulbaire » (un cinquième des cas[3]), débutant dans les territoires d'innervation bulbaires (déglutition, phonation et motricité de la langue)[2].
Toutefois, l'évolution des atteintes tend à se généraliser à l'ensemble des territoires moteurs et la distinction entre ces deux types n'est pas toujours évidente lors du diagnostic. Les femmes présentent statistiquement plus de formes bulbaires que les hommes[4].
Symptômes
Quels que soient les territoires concernés, la symptomatologie initiale se manifeste par la coexistence de troubles moteurs centraux (syndrome pyramidal : hyperréflexie, spasticité) et d'atteintes neurogènes périphériques - crampes, perte de force musculaire, fonte musculaire (« amyotrophie »). Les fasciculations sont rares, mais très spécifiques, surtout si elles concernent la langue[5]. Il n'existe aucun trouble sensitif dans cette maladie.
Un syndrome pseudo-bulbaire (rires et pleurs spasmodiques) peut survenir. Une atteinte des fonctions cognitives, de type dégénérescence fronto-temporale (DFT), se rencontre parfois. Dans certaines formes héréditaires, un membre de la famille peut développer soit une dégénérescence lobaire fronto-temporale, soit une SLA.
Évolution
La SLA cause une paralysie progressive accompagnée d'une amyotrophie. L'atteinte de la fonction respiratoire est le principal facteur du pronostic. L'évolution se fait toujours vers l'aggravation des déficits, mais à une vitesse variable. Ainsi, la durée totale de la maladie, c'est-à-dire l'intervalle entre l'apparition du premier symptôme et le décès, peut varier de quelques mois à plusieurs années ; elle est en moyenne inférieure à quatre ans et dans 50 % des cas le décès a lieu dans les trois ans suivant la première manifestation clinique[6]. Des survies prolongées sont cependant possibles conduisant, dans les formes évoluées, à une perte d’autonomie et à une dégradation de l'état général. L'apparition de troubles de la déglutition favorise la dénutrition. L'atteinte des muscles respiratoires favorise les infections, et peut conduire à une insuffisance respiratoire nécessitant une ventilation mécanique. L'atteinte de la parole rend la communication difficile.
Épidémiologie
L'incidence de la maladie est d'environ 2 pour 100 000 par an. Sa prévalence est de 5 à 7 pour 100 000[3]. Aux États-Unis, une étude basée sur l'analyse de publications scientifiques consacrées à la SLA, a relevé : « L'incidence de la SLA peut être plus faible chez les Africains, Asiatiques, et ethnies hispaniques que chez [les personnes blanches non hispaniques]. Nous concluons avec des propositions pour une étude épidémiologique prospective se concentrant sur les populations non caucasiennes »[7].
Une augmentation globale de l'incidence, d'environ 50 % sur les 50 dernières années[Depuis quand ?], est attribuée d'une part à l'augmentation de l'espérance de vie des populations, d'autre part à de meilleurs critères diagnostiques. Cependant, une participation environnementale n'est pas exclue. Certaines localités du Pacifique sont connues pour avoir une très forte sur-incidence de SLA (île américaine de Guam, péninsule japonaise de Kii), associées à des syndromes démentiels et à la maladie de Parkinson. Elles ont engendré de nombreuses études, mais les intrications entre génotype, environnement et mode de vie ne sont toujours pas élucidées, pas plus que ne l'est le lien entre la SLA et les autres pathologies neurodégénératives.
Le pic d'incidence se situe entre 55 et 70 ans. Des personnes âgées de plus de 80 ans peuvent être atteintes[8]. Le rapport hommes/femmes est entre 1,3 et 2 (il tend à s'égaliser)[6]. C'est la maladie du motoneurone la plus fréquente chez l'adulte[9].
Enfin, il faut distinguer la forme sporadique (distribution au hasard dans la population) et la forme familiale. La première forme est observée dans 90 à 95 % des cas. La forme héréditaire correspond à 5 à 10 % des cas[10], la plupart étant de forme autosomique dominante[3]. Les premiers signes d'une SLA héréditaire surviennent statistiquement plus tôt (46 ans en moyenne) que les formes sporadiques (56 ans en moyenne).
Causes
Jusqu'en 2011, plusieurs mécanismes étaient soupçonnés d'expliquer l'atteinte spécifique des motoneurones sans qu'aucun n'ait été formellement établi. Plusieurs pistes sont explorées :
- une dérégulation cellulaire de la gestion du stress oxydatif, comme le montre l'implication des gènes codant la superoxyde dismutase (SOD) dans les formes familiales ;
- un phénomène d'excito-toxicité : soit par excès de glutamate ou d'une molécule apparentée (par exemple l'aspartame), soit par des mutations modifiant les récepteurs au glutamate ;
- une dérégulation des mécanismes d'apoptose (mort programmée de la cellule)[11] ;
- une polymérisation désordonnée des neurofilaments entraînant une perturbation du transport des vésicules.[évasif][réf. souhaitée]
Depuis, les causes sont attribuées à un ensemble de mutations génétiques et de dysfonctionnements cellulaires, impliquant notamment les protéines FUS, TDP-43 (en), l'ubiquiline 2 (UBQLN2) et C9ORF72[12],[13].
SLA sporadiques
Les SLA sporadiques concernent 90 % des personnes atteintes par cette maladie. Si de nombreuses études ont été réalisées à ce jour, aucun facteur de risque ne peut être retenu avec suffisamment de certitude pour permettre d'en affirmer la causalité. Parmi ceux les plus fréquemment cités on trouve les facteurs environnementaux suivants :
- le métier d'agriculteur et/ou l'exposition aux pesticides. Plusieurs études scientifiques[14],[15],[16],[17] ont trouvé des corrélations statistiques entre certains pesticides agricoles et la SLA[source secondaire nécessaire] ;
- les traumatismes physiques importants et/ou le sport à haut niveau[18],[19],[20] ;
- l'exposition aux métaux lourds (hypothèse très discutée) ;
- le tabagisme[21] ;
- l'entérovirus (cousin du poliovirus), dont une réactivation tardive expliquerait la maladie (plusieurs cas de SLA régressives après régénérescence de l'immunité ont été décrits chez des patients atteints du SIDA) ;
- une intoxication alimentaire par un acide aminé semblable au glutamate, la BMAA (bêta-N-méthylamino-L-alanine), qui est produite par la plupart des cyanobactéries, et dont l'ingestion à hautes doses chez des primates provoque un ensemble de neurodégénérescences compatibles avec la SLA ;
- le rôle de l'alimentation est encore discuté[12]. En Savoie, après un nombre anormal de cas dans le village de Montchavin, une équipe de chercheurs évoque un lien entre le développement de la SLA et la consommation d'un champignon : le gyromitre géant[22],[23] ou fausses morilles. En 2021, dans le Journal of the Neurological Sciences (en), les chercheurs rapportent qu'après avoir éliminé les autres causes possibles (toxines bactériennes, plomb dans l'eau, gaz radon dans les maisons…), il avait été constaté que les 14 personnes atteintes, sans lien de parenté mais se connaissant, avaient bien consommé et à plusieurs reprises sur plusieurs années ce champignon, contrairement aux autres villageois[24]. Une augmentation des cas de SLA a aussi été constaté dans une région de Finlande où ce champignon est très consommé[24]. Sur l'île de Guam, la graine de Cycas du Japon consommé traditionnellement s'est révélé être à l'origine de nombreux cas de SLA[24].
La plupart de ces facteurs de risque ont été mis en évidence grâce à des études cas-témoins basées sur de très faibles effectifs. Suivant l'hypothèse d'une exposition environnementale, les anciens combattants de la guerre du Golfe atteints de SLA sont indemnisés. L'existence de gènes de susceptibilité pour la SLA est également suspectée[25]. Il est cependant difficile de faire la part des choses entre cas sporadiques, cas héréditaires à pénétrance incomplète et terrain de susceptibilité génétique, d'autant plus que les gènes responsables des formes familiales reconnues comme telles ne sont, pour la plupart, pas encore isolés.
SLA génétiques ou familiales
De 5 à 10 % des cas de SLA pourraient avoir une cause génétique[5]. Les cas génétiques certains sont classés en fonction du mode de transmission et du gène impliqué.
La transmission autosomique dominante est la plus fréquente. Des mutations sur le gène SOD1 d'une dismutase, située sur le chromosome 21 ont été détectées dans 10 à 20 % des cas familiaux de la maladie, mais également dans quelques cas sporadiques[26]. Une mutation sur le gène C9orf72 peut être également à l'origine de cas familiaux.
La transmission autosomique récessive est plus rare et concerne surtout certaines populations de l'Afrique du Nord[27]. Elle implique trois gènes différents :
| Fréquence | Gène | Chromosome | Protéine |
|---|---|---|---|
| Rare | ALS2 | 15q15.1-q21.1 | Alsine |
| Rare | 2q33 | ||
| Rare | SPG20 | 13q12.3 | Spartine |
Les autres maladies en rapport avec une mutation pathologique du gène ALS2 comprennent : la sclérose latérale primitive, la paraplégie spastique familiale ascendante à début précoce et une forme particulière de sclérose latérale amyotrophique la sclérose latérale amyotrophique juvénile. Une mutation du gène SPG20 codant la protéine spartine est aussi responsable du syndrome de Troyer.
Une forme génétique de SLA avec transmission liée à l'X a été identifiée dans une famille[28].
Une mutation du gène UBQL2 codant la protéine ubiquiline 2 est responsable de la dégénérescence motoneuronale dans certaines formes de SLA familiales[29]. Cette protéine, dont la fonction normale est de réparer les cellules endommagées dans les neurones moteurs et corticaux, entraîne quand elle est défectueuse l'accumulation de cette protéine et de celles qui auraient dû être réparées.
En 2011, une anomalie génétique consistant en la répétition d’un hexanucléotide a été identifiée dans une région du chromosome 9 nommée C9ORF72 ; celle-ci est responsable d’une forme de SLA associée à une démence fronto-temporale (DFT)[30] et serait également à l’origine de 6 % de tous les cas de SLA parmi les européens caucasiens[31]. Le taux élevé de mutations découvertes dans des cas apparemment sporadiques (c’est-à-dire sans histoire familiale) suggère que les anomalies génétiques pourraient jouer un rôle plus important que ce que l’on pensait jusqu’ici, alors que l’exposition à des facteurs de l’environnement serait moins déterminante.
Diagnostic
Signes cliniques
Des critères de diagnostic stricts ont été établis principalement pour la recherche[32] : preuves évidentes cliniques, électriques ou anatomo-pathologiques d'atteinte du neurone moteur périphérique et preuves cliniques d'atteinte du neurone moteur central et apparition progressive de signe clinique dans d'autres régions avec l'absence de signes électriques ou anatomo-pathologiques pouvant expliquer une atteinte du neurone moteur périphérique et/ou central, et une imagerie cérébrale ne permettant pas d'expliquer les signes cliniques ou électriques.
L'examen neurologique doit mettre en évidence des signes d'atteinte du neurone moteur périphérique et/ou central dans au moins quatre régions : tronc cérébral, cervical, thoracique et lombaire. Parmi les signes cliniques, l'aspect des mains en mains de singe est constaté, correspondant à une amyotrophie des muscles interosseux, sans que l'agilité soit altérée. Il n'y a classiquement pas de troubles cognitifs. Il existe cependant des déficits inconstants dans la fluence verbale et le raisonnement[33]. Des formes plus évoluées entraînent des signes ressemblant à ceux d'une dégénéréscence fronto-pariétale[34]. Cette similarité n'est pas qu'anecdotique : certaines lésions étant très semblables[35] et les deux maladies ayant une certaine ressemblance génétique et en imagerie fonctionnelle, l'hypothèse que certaines formes soient apparentées est évoquée[5].
Du fait du début insidieux des symptômes, le diagnostic est souvent retardé, le délai dépassant fréquemment un an après les premiers signes de la maladie[36].
Il faut penser à une polymyosite chez un adulte jeune présentant les signes de SLA mais dont l'atrophie débute par des muscles proximaux.
L'apparition de fasciculations diffuses et ceci depuis plus de 2 mois sans réelle faiblesse exclut la SLA, a fortiori chez des adultes jeunes (moins de 30 ans).
Examens complémentaires
La biopsie musculaire montre une atrophie des cellules musculaires avec des lésions relativement spécifiques[37]. L'électromyographie montre, dans les formes évoluées, une diminution de l'amplitude de la conduction nerveuse évocatrice d'une dénervation[38]. À l'étude, la vitesse de conduction des nerfs sensoriels reste normale. Le tracé électromyographique montre, lui-même, des anomalies spécifiques[39]. Il permet en général de retrouver un syndrome neurogène périphérique diffus, sans ralentissement des vitesses de conduction ni bloc proximal. Il confirme la clinique, détecte des atteintes infracliniques et écarte d'autres neuropathies périphériques. L'IRM permet essentiellement d'éliminer les autres diagnostics, dont les atteintes mécaniques de la moelle épinière.
Sont également notés l'analyse du liquide céphalo-rachidien par ponction lombaire (normale), les CPK sont augmentées, les enzymes hépatiques sont perturbées et le scanner montre une absence de compression médullaire.
Traitement
Traitement médicamenteux
Deux traitements médicamenteux sont présentement approuvés pour le traitement de la SLA, soit le riluzole et l'édaravone. Le riluzole, étudié depuis 1996, a une action modérée avec une prolongation de la vie de l'ordre de quelques mois[40]. L'édaravone, approuvé aux États-Unis depuis 2017, a une efficacité modérée puisqu'elle permet de ralentir l'évolution de la maladie par environ 33 % chez les patients dans la phase initiale de la maladie[41].
Le masitinib est en cours de demande d'Autorisation de mise sur le marché en Europe[42], l'Agence européenne des médicaments lui ayant préalablement accordé le statut de médicament orphelin[43]. L'autorisation de mise sur le marché a été refusée une première fois en avril 2018, mais le laboratoire a fait appel[44], avant d'y renoncer. Cette première demande était basée sur 50 % des patients de l'étude et l'agence européenne considérait que les données cliniques n'étaient pas assez « robustes ». La société prévoit de faire une nouvelle demande d'AMM en 2019 sur la base de 100 % des patients en plus d'une étude complémentaire avec un dosage plus fort.
Prometteuse, la molécule Tirasemtiv avait atteint les tests de phase 3, avant d'être arrêtés en novembre 2018 faute d'efficacité[45]. Une molécule apparentée avec un meilleur profil d'effets secondaire, le Reldesemtiv, est présentement à l'essai[46].
Traitement non pharmacologique et symptomatique
Le traitement est donc essentiellement symptomatique et inclut kinésithérapie (appareillage spécifique pour ralentir la progression du déficit moteur) et ergothérapie pour aider à pallier les difficultés dans les activités de la vie quotidienne, orthophonie pour les troubles dysarthriques (troubles moteurs affectant la réalisation de la parole) et de déglutition, lutte contre les infections bronchiques par antibiothérapie (afin d'éviter l'aggravation d'une altération de la fonction respiratoire), traitement médicamenteux symptomatique des crampes et de la spasticité liée au syndrome pyramidal, prise en charge des problèmes nutritionnels pouvant aller jusqu'à une gastrostomie en cas de troubles sévères de la déglutition, voire pose d'une sonde gastrique à demeure, ventilation mécanique en cas d'atteinte du muscle de la respiration, le diaphragme. La mise en place d'une ventilation non invasive (par masque) permet un confort plus important et une durée de vie substantiellement allongée[47], et la prise en charge du syndrome dépressif fréquent. Des équipements permettent de faciliter la vie de tous les jours, en particulier, des interfaces avec des ordinateurs ou des tablettes graphiques[48].
À titre anecdotique, il est noté que le Dr Murray Sanders proposa après guerre un traitement à base de venin de cobra[49][source insuffisante].
Personnalités atteintes par la maladie
Artistes
Les musiciens Charlie Mingus, Luc Cousineau et Leadbelly, le producteur de rap Pone[50] membre de la Fonky Family qui a d'ailleurs créé un site web dédié à cette maladie[51], le chanteur Frank Alamo[52], l'acteur David Niven, l'acteur Frederick Weibgen et le guitariste Jason Becker sont ou ont été atteints de la maladie.
L'artiste allemand Jörg Immendorff en meurt en 2007, l'acteur vénézuélien Mariano Álvarez Fernández en meurt en 2001. Dan Toler, le guitariste des Former Allman Brothers Band succombe en février 2013 après avoir lutté pendant deux ans[53]. Le bassiste Mike Porcaro en est mort en mars 2015[54]. L'acteur et écrivain Sam Shepard meurt des suites de cette maladie en 2017[55] ainsi que l'acteur, metteur en scène, dramaturge et directeur de théâtre français Adel Hakim[56]. Le 2 octobre 2019, la frontwoman du groupe californien The Muffs et auteure/chanteuse/guitariste/bassiste de nombreux autres groupes, Kim Shattuck, meurt dans son sommeil des suites de la sclérose latérale amyotrophique diagnostiquée deux ans auparavant[57]. Le compositeur et interprète finlandais Pave Maijanen (en), qui avait représenté la Finlande à l'Eurovision en 1992 meurt le de la maladie, laquelle lui avait été diagnostiquée en 2018[58].
Le dessinateur et scénariste Stephen Hillenburg, créateur de Bob l'éponge, est décédé le 26 novembre 2018 de la maladie de Charcot.
L'humoriste Jean-Yves Lafesse meurt le de cette maladie[59].
Écrivains et historiens
C'est le cas des écrivaines françaises Marcelle Delpastre, Florence Bouhier, Anne Bert, des écrivains Bernard Lenteric et Frédéric Badré, de l'historien de l'art Daniel Arasse et de Matthieu Galey[60], critique littéraire et dramatique. Tony Judt, historien atteint de la maladie depuis 2008, en meurt en 2010[61]. Ariel Crozon en tire un récit, L'Affreuse[62],[63]. Marie Sey décrit son combat de femme dans la tourmente de la SLA dans Vivre jusqu'à en mourir, préfacé par le Professeur Axel Kahn, en 2019[64].
Scientifiques

Le physicien Stephen Hawking, le mathématicien Fokko du Cloux[66], le médecin Roy Walford en étaient atteints.
Personnalités politiques
- Mao Zedong pourrait également être mort de cette maladie, à l'âge de 82 ans[67],[68].
- Friedrich Paulus (1890-1957), Generalfeldmarschall du Troisième Reich.
- Yasuhiko Funago (1957-), homme politique japonais du Reiwa Shinsengumi.
Sportifs
Le joueur de baseball américain Lou Gehrig en meurt en 1941. En 1996, le footballeur belge Claude Bissot meurt à 49 ans de la SLA[69]. En 2006, la maladie emporte l'ancien joueur écossais Jimmy Johnstone à l'âge de 61 ans[70]. C'est le 30 décembre 2010 que l'ancien joueur de football canadien, Tony Proudfoot[71], meurt, moins de quatre ans après avoir été diagnostiqué. Atteint également par cette maladie, l'ancien footballeur de la Fiorentina et du Milan AC Stefano Borgonovo[72] en est mort en 2013. Ce dernier avait créé la « fondation Stefano Borgonovo » qui contribue pour la recherche sur la SLA. Le 30 octobre 2013 Fernando Ricksen le footballeur néerlandais a révélé sa maladie lors de l'émission télévisée néerlandaise De Wereld Draait Door[73]. ll meurt le 18 septembre 2019[74].
L'ancien basketteur Sherron Mills est également décédé [75]de cette maladie[76].
Le triathlète Ironman Jonathan "Blazeman" Blais le fondateur de The Blazeman Foundation for ALS, en est mort en 2007, deux ans après avoir été diagnostiqué[77]. Joost van der Westhuizen, rugbyman sud-africain est décédé de la maladie le 6 février 2017 à l'âge de 45 ans[78].
Le 1er août 2017, l'ancien joueur de tennis français Jérôme Golmard, qui avait été diagnostiqué en 2014, succombe aux suites de cette maladie à l'âge de 43 ans[79]. Le joueur de tennis Brad Drewett en était mort en 2013[80].
L'athlète français Frédéric Dahmani, disparu à 48 ans le 31 décembre 2018, avait écrit un livre sur son cas, intitulé Quand Charcot s'invite au trail…[81].
Le 29 novembre 2020, le joueur de football emblématique du Sénégal, et buteur contre l'équipe de France lors du mondial 2002, Papa Bouba Diop, meurt de cette maladie à l'âge de 42 ans[82].
Tissu associatif
En France, les malades sont représentés et soutenus par plusieurs associations, dont l'Association pour la recherche sur la SLA (ARSLA[83]) ou la fondation Thierry-Latran[84] (sous l'égide de la Fondation de France), qui financent également la recherche. Un ensemble d'associations créées chacune autour d'un malade en particulier se sont regroupés depuis 2017 sous l’appellation Collectif Solidarité Charcot[85]. D’autres petites associations existent comme les p’tites mains de Charcot qui forment des auxiliaires de vie pour la prise en charge des personnes SLA à domicile ou Tous En selles Contre la SLA qui a pour objectif de faire avancer la recherche contre la SLA en favorisant l’émergence d’études sur l’axe intestin/cerveau.
Historique


La maladie a été étudiée par Guillaume Duchenne de Boulogne dans les années 1850, qui pense que celle-ci n'est que musculaire. La première démonstration d'une atteinte de la moelle épinière est faite en 1853 par Cruveilhier au cours d'une autopsie, montrant une atrophie des racines antérieures. En 1860, Duchenne décrit cliniquement la forme bulbaire de la maladie et Jean-Martin Charcot démontre l'atteinte du bulbe dans ce cas. Ce dernier publie plusieurs cas d'atrophies musculaires et d'atteinte médullaire. En 1874, il présente ses conclusions dans ses douzième et treizième leçons, décrivant avec précision les symptômes et les lésions macroscopiques et microscopiques de la moelle[86].
Cas groupés
Indices de facteurs professionnels et environnementaux
Une étude cas-témoins italienne publiée en 2010 a conclu qu'une activité professionnelle associée à une forte activité physique est un facteur de risque, mais que les traumatismes sportifs ne sont pas directement un facteur de risque de SLA[87].
La littérature scientifique rapporte quelques cas de couples touchés par la maladie (phénomène statistiquement très improbable)[88]. D'autres cas groupés qui pourraient laisser penser qu'une « contagion » est, dans certains cas, possible, ou qu'une « cause environnementale » commune puisse parfois exister.
Par exemple, dans un même immeuble de Chicago, ce sont 11 patients qui meurent de cette maladie, sans explication trouvée, ou encore chez des vétérans parmi le personnel déployé sur le terrain lors de la guerre du Golfe[89],[90],[91],[92] par rapport au personnel non déployé (avec des chiffres cependant méthodologiquement discutés[93]).
De même, une épidémie semble spécifiquement concerner les footballeurs italiens ou ayant pratiqué en Italie[94].
Le cas des footballeurs italiens
C'est le cas de spécificité professionnelle qui a été le plus médiatisé et le plus flagrant, il fait l'objet d'une enquête judiciaire depuis plusieurs années. Ces anciens footballeurs professionnels italiens sont sept à huit fois plus touchés que le reste de la population[95]. Près de 40 morts[réf. nécessaire] de 1973 à 2008 (parmi au moins 51 victimes de la maladie détectées chez 30 000 footballeurs ayant joué en Italie de 1950 à 2005 environ). La première victime a été Armando Segato (milieu de terrain de la Fiorentina (1952-1960), mort à 43 ans d'une sclérose latérale amyotrophique (en 1973), puis près de 40 footballeurs célèbres du championnat d'Italie de football ont suivi dont Guido Vincenzi (Sampdoria, 1958-1969), Narciso Soldan (Torino Football Club, 1959-1961), Giorgio Rognoni (Milan AC, 1967-1971), Giuseppe Longoni (Fiorentina, 1969-1973), Adriano Lombardi (Avellino, 1975-1979), Rino Gritti (Avellino, 1975-1977), Albano Canazza (Côme, 1980-1983), Fabrizio Dipietropaolo (AS Rome, 1981-1983), Fabrizio Falco (Salernitana, 1985-1989), Gianluca Signorini (Parme, 1960-2002)[96], Lauro Minghelli (Torino, 1989-1993), Ubaldo Nanni (Pise, 1992-1994). Plusieurs joueurs phares des années 1980 sont aussi touchés, Stefano Borgonovo notamment.
À la fin du XXe siècle, le procureur Guariniello découvre ce fait alors qu'il enquêtait sur le dopage dans les clubs de Série A (première division). Il constate aussi un nombre anormalement élevé de pathologies cardiovasculaires et de cancers (pour 24 000 joueurs étudiés, le nombre de cancers était le double du nombre attendu, avec 13 cas de cancer du colon, 9 de cancer du foie, 10 cas de cancer du pancréas au lieu de respectivement 6, 4 et 5 cas en termes de risque statistique. De plus, alors que la SLA ne conduit à la mort qu'après plus de deux ans chez 50 % des patients « normaux », et que 10 à 20 % des patients survivent plus de 10 ans[97], les footballeurs italiens en sont presque tous morts très rapidement (en quelques mois).
La justice italienne (parquet de Turin) a demandé à un épidémiologiste (le Pr Adriano Chio, neurologue) d'étudier un panel de 7 325 ex-footballeurs en activité entre 1970 et 2006, Les professionnels présentaient également un risque près de 7 fois plus élevé que la normale de développer la maladie, alors que les footballeurs-amateurs ne sont pas touchés. Pour le cas des 5 footballeurs professionnels nés en Italie et ayant joué entre 1970 et 2001 morts de SLA, le risque était 20 fois supérieur au taux normal[98]. Rien ne permet à ce stade de savoir s'il s'agit d'un empoisonnement ou si une cause microbienne pourrait être en cause (des cas groupés de maladie nosocomiale ont été observés aux États-Unis). Curieusement, l'étude révèle que la plupart des malades occupaient le poste de milieu de terrain (plus précisément, « Le risque de la SLA a été plus élevé pour les carrières durables de 5 ans, pour les milieux de terrain, et pour les joueurs engagés après 1980 »[94]).
Une information judiciaire contre X pour homicide involontaire est ouverte en 1998 par le procureur qui envisage trois pistes :
- L'impact d'un dopant qui aurait pu n'être utilisé qu'en Italie chez les footballeurs ; les corticoïdes et anabolisants sont maintenant connus comme causes possibles de symptômes de ce type[95] ;
- Des traumatismes du cerveau ou de la moelle épinière liés au football de haut niveau (ils étaient suspectés), mais outre qu'on ne trouve hors d'Italie qu'un seul footballeur touché par la SLA (Jimmy Johnstone, ex-attaquant du Celtic FC, vedette des années 1960), une étude statistique poussée avec cas-témoins n'a pas pu confirmer que ce type de traumatisme était un facteur de risque (alors qu'une hyperactivité professionnelle l'était)[87] ; de même une enquête commandée par le procureur Guariniello, rendue en septembre 2008, n'a mis aucun cas semblable en évidence dans le cyclisme (pour 1 701 coureurs professionnels masculins ayant participé à des championnats entre 1945 et 2001) ni le basket (pour 7 325 joueurs professionnels masculins ayant joué des championnats entre 1980 et 2004)[94] ;
- Les traitements pesticides utilisés pour l'entretien des pelouses sportives.
Notes et références
- Voir sur lougehrig.com.
- « SLA : mon corps est une prison », 36.9°, Radio télévision suisse, (résumé, lire en ligne [vidéo]), présentation : Isabelle Moncada
- McDermott CJ, Diagnosis and management of motor neurone disease, Shaw PJ, BMJ, 2008;336:658-662
- la maladie de Charcot ou sclérose latérale amyotrophique, paragraphe "Quelles sont les manifestations de la SLA ?" sur frcneurodon.org, consulté le 3 octobre 2019.
- Kiernan MC, Vucic S, Cheah BC et al. Amyotrophic lateral sclerosis, Lancet, 2011;377:942-955
- Mitchell JD, Borasio GD, Amyotrophic lateral sclerosis, Lancet, 2007;369:2031-2041
- (en) S Cronin, O Hardiman et BJ Traynor, « Ethnic variation in the incidence of ALS: a systematic review », Neurology, no 68, , p. 1002-1007 (lire en ligne)
- Jelena Grujic, Martial Coutaz et Jérôme Morisod, « La sclérose latérale amyotrophique atteint aussi le quatrième âge (Amyotrophic lateral sclerosis also threatens the octogenarian) », Rev med suisse, vol. 4, no 159, , p. 1353-1357 (PMID 18592728, lire en ligne, consulté le )
- « La maladie de Charcot (SLA) : causes, symptomes, traitements, recherches - ICM », sur Institut du Cerveau (consulté le )
- (en) C Lomen Hoerth, J Murphy, S Langmore, JH Kramer, RK Olney et B Millner, « Are amyotrophic lateral sclerosis patients cognitively normal », Neurology, no 60, , p. 1094-1097
- Dr Hélène Blasco, « La recherche », sur ARSLA (consulté le )
- (en) Albert C. Ludolph, Johannes Brettschneider et Jochen H. Weishaupt, « Amyotrophic lateral sclerosis: », Current Opinion in Neurology, vol. 25, no 5, , p. 530–535 (ISSN 1350-7540, DOI 10.1097/WCO.0b013e328356d328, lire en ligne, consulté le )
- Michele Longoni Calió, Elisandra Henriques, Amanda Siena et Clélia Rejane Antonio Bertoncini, « Mitochondrial Dysfunction, Neurogenesis, and Epigenetics: Putative Implications for Amyotrophic Lateral Sclerosis Neurodegeneration and Treatment », Frontiers in Neuroscience, vol. 14, , p. 679 (ISSN 1662-453X, PMID 32760239, PMCID PMC7373761, DOI 10.3389/fnins.2020.00679, lire en ligne, consulté le )
- (en) F Bonvicini, N Marcello, J Mandrioli, V Pietrini et M Vinceti, « Exposure to pesticides and risk of amyotrophic lateral sclerosis: a population-based case-control study », Ann Ist Super Sanita, vol. 46, no 3, , p. 284-287 (PMID 20847462)
- (en) AM Malek, A Barchowsky, R Bowser, A Youk et EO Talbott, « Pesticide exposure as a risk factor for amyotrophic lateral sclerosis: A meta-analysis of epidemiological studies: Pesticide exposure as a risk factor for ALS », Environ Res., no 117, , p. 112-119 (PMID 22819005)
- (en) M Vinceti, I Bottecchi, A Fan, Y Finkelstein et J Mandrioli, « Are environmental exposures to selenium, heavy metals, and pesticides risk factors for amyotrophic lateral sclerosis? », Rev Environ Health, vol. 27, no 1, , p. 19-41 (PMID 22755265)
- (en) F Kamel, DM Umbach, RS Bedlack, M Richards, M Watson et al., « Pesticide exposure and amyotrophic lateral sclerosis », Neurotoxicology, vol. 33, no 3, , p. 457-462 (PMID 22521219)
- (en) CA Harwood, CJ McDermott et PJ Shaw, « Physical activity as an exogenous risk factor in motor neuron disease (MND): a review of the evidence », Amyotroph Lateral Scler, no 10, , p. 191-204 (PMID 19263258)
- A Drouet et al., « Sclérose latérale amyotrophique (SLA) chez les militaires français : enquête rétrospective », Revue neurologique, no 166, , p. 621-629
- (en) M Gotkine, Y Friedlander et Hochner, « Triathletes are over-represented in a population of patients with ALS », Amyotroph Lateral Sclerosis and Frontotemporal Degeneration, , p. 1-3 (PMID 25007701)
- (en) V Gallo, HB Bueno-De-Mesquita, R Vermeulen et al., « Smoking and risk for amyotrophic lateral sclerosis: analysis of the EPIC cohort », Ann Neurol, no 65, , p. 378-385 (lire en ligne)
- « Un champignon à l’origine de cas de SLA/Maladie de Charcot », sur La Voix du Nord, (consulté le )
- Santé Publique France, « Rapport d'investigation de suspicions d’agrégats de scléroses latérales amyotrophiques (SLA) en Savoie et en Isère » [PDF],
- Pierre Kaldy, « Un champignon lié à des cas de maladie de Charcot », Sciences et avenir, no 895, , p. 62
- (en) MA van Es, JH Veldink, CG Saris et al., « Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis », Nat Genet, no 41, , p. 1083-1087 (lire en ligne)
- Rosen DR, Siddique T, Patterson D et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis, Nature, 1993; 362: 59-62
- Figlewicz DA, Orrell RW, The genetics of motor neuron diseases, Amyotroph Lateral Scler Other Motor Neuron Disord 2003;4:225–231
- Hong S, Brooks BR, Hung WY, et al (1998) X-linked dominant locus for late-onset familial amyotrophic lateral sclerosis. Abstr Soc Neurosci 24:478
- (en) Deng et al., Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia, dans Nature (2011) DOI:10.1038/nature10353
- Dejesus-Hernandez, M., et al.,, « Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. », Neuron, vol. 72, no 2, , p. 245–56 (PMID 21944778, DOI 10.1016/j.neuron.2011.09.011, lire en ligne)
- Majounie E., et al., « Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. », Lancet Neurology, vol. 11, no 4, , p. 323–330 (DOI 10.1016/S1474-4422(12)70043-1)
- Brooks BR, Miller RG, Swash M, Munsat TL (2000) El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1:293-9
- Flaherty-Craig C, Eslinger P, Stephens B, Simmons Z, A rapid screening battery to identify frontal dysfunction in patients with ALS, Neurology, 2006;67:2070-2072
- Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE, Prevalence and patterns of cognitive impairment in sporadic ALS, Neurology, 2005;65:586-590
- Neumann M, Sampathu DM, Kwong LK et Als. Ubiquitinated TDP-43 in fronto-temporal lobar degeneration and amyotrophic lateral sclerosis, Science, 2006;314:130-133
- Chio A, ISIS Survey: an international study on the diagnostic process and its implications in amyotrophic lateral sclerosis, J Neurol, 1999;246(suppl 3):III1-III5
- Baloh RH, Rakowicz W, Gardner R, Pestronk A, Frequent atrophic groups with mixed-type myofibers is distinctive to motor neuron syndromes, Muscle Nerve, 2007;36:107-110
- Daube JR, Electrodiagnostic studies in amyotrophic lateral sclerosis and other motor neuron disorders, Muscle Nerve, 2000;23:1488-1502
- de Carvalho M, Dengler R, Eisen A et Als. Electrodiagnostic criteria for diagnosis of ALS, Clin Neurophysiol, 2008;119:497-503
- Miller RG, Mitchell JD, Lyon M, Moore DH Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND), Cochrane Database Syst Rev 2007(1):CD001447
- Writing Group et Edaravone (MCI-186) ALS 19 Study Group, « Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial », The Lancet. Neurology, vol. 16, no 7, , p. 505–512 (ISSN 1474-4465, PMID 28522181, DOI 10.1016/S1474-4422(17)30115-1, lire en ligne, consulté le )
- « Dépôt d'AMM pour la SLA »,
- « Médicament orphelin contre la SLA »
- (en) « Agence Européenne des Médicaments - Refus de l'autorisation de mise sur le marché du masitinib », sur www.ema.europa.eu, (consulté le )
- (en-US) « Tirasemtiv », sur ALS News Today (consulté le )
- (en) « Cytokinetics Announces Results of FORTITUDE-ALS, a Phase 2 Clinical Trial of Reldesemtiv in Patients With ALS, Presented at American Academy of Neurology Annual Meeting », sur Cytokinetics, Inc. (consulté le )
- Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ, Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial, Lancet Neurology, 2006;5:140-7
- « Une application pour aider les patients atteints de la maladie de Charcot », sur infoDSI, (consulté le ).
- (en) [PDF] « The Historical Use of Snakes in Medicine and Why We’re Studying Cobra Venom as a Treatment for Multiple Sclerosis and other Disorders », (consulté le )
- « La Fonky Family : une reformation pour la bonne cause » sur mouv.fr.
- « La Sla pour les nuls | Un des symptômes de la SLA étant les troubles musculaires (crampes, fasciculations, spasticité etc.) », sur laslapourlesnuls.com (consulté le )
- Avec AFP, « Frank Alamo, l'interprète de "Ma biche", est mort », Le Monde.fr, (lire en ligne).
- Voir sur guitarworld.com.
- « Mike Porcaro est mort : l'ancien bassiste de Toto est décédé des suites de la maladie de Charcot », sur huffingtonpost.fr, (consulté le ).
- Charles Decant, « L'acteur Sam Shepard est mort » sur PureMédias, 31 juillet 2017.
- Voir sur lemonde.fr.
- (en) Kim Shattuck, Muffs Founder and Singer, Dead at 56, Suzy Exposito & Althea Legaspi, RollingStone, 2 octobre 2019.
- (en-GB) « 🇫🇮 Pave Maijanen passes away, aged 70 », sur ESCXTRA.com, (consulté le ).
- « L’humoriste Jean-Yves Lafesse est décédé », sur LA VDN, (consulté le ).
- Jean-Martial Gane, Un écrivain face à sa mort prochaine : la sclérose latérale amyotrophique vue à travers le journal de Matthieu Galey, 1989.
- (en) Tony Judt: A Public Intellectual Remembered, Michael Elliott, Time, 7 août 2010.
- https://www.babelio.com/auteur/Ariel-Crozon/434034
- « La vie, quand même - La République des livres », sur La République des livres, (consulté le ).
- « Marie Sey: Livres, bibliographie de l'auteur - Éditions Eyrolles », sur www.editions-eyrolles.com (consulté le )
- (en) « From charcot to lou gehrig: deciphering selective motor neuron death in als : Abstract : Nature Reviews Neuroscience. »
- Famous People with Amyotrophic lateral sclerosis Lou Gehrigs Disease sur Disabled world, 13 mars 2008.
- (en) Zhisui Li, Private Life Of Chairman Mao : The Memoirs of Mao's Personal Physician (illustrated, reprint ed.), Random House, , 720 p. (ISBN 978-1-4070-5922-8, lire en ligne), p. 581
- « Comment meurent les dictateurs » sur tempsreel.nouvelobs.com.
- « Une journée pour la sclérose latérale amyotrophique », 7 sur 7, 18 juin 2009.
- Voir sur jimmy.johnstone.muchloved.com.
- (en) « My time has come: Tony Proudfoot. »
- « La lutte de Stefano Borgonovo contre la SLA » sur handi-run.sport24.com.
- Voir sur dailymail.co.uk.
- « L’ancien footballeur néerlandais Fernando Ricksen est décédé à l’âge de 43 ans », sur Le Soir, (consulté le )
- « DÉCÈS DE SHERRON MILLS - BCM », (consulté le )
- (en-US) « Help Sherron Mills’ Fight With ALS », sur Ram Nation, (consulté le )
- Voir sur waronals.com.
- « Le rugbyman sud-africain Joost van der Westhuizen, champion du monde en 1995, est mort. », sur lemonde.fr, (consulté le ).
- « Tennis : l'ancien no 1 français Jérôme Golmard est mort à l'âge de 43 ans » sur europe1.com, 1er août 2017.
- Mort de Brad Drewett, président de l'ATP, L'Équipe, 3 mai 2013.
- Voir sur rebeyl.com/dahmani
- « L'ancien international sénégalais Papa Bouba Diop est mort à 42 ans », sur L'Équipe (consulté le )
- Site de l’ARSLA
- Site de la fondation.
- Voir sur le site de la fondation Thierry-Latran.
- Dupont JC, La fondation de la neurologie, Les Génies de la science, 2008;7:42-51
- E. Beghi, G. Logroscino, A. Chiò, O. Hardiman, A. Millul, D. Mitchell, R. Swingler et B.J. Traynor, « Amyotrophic lateral sclerosis, physical exercise, trauma and sports: Results of a population-based pilot case-control study », Amyotrophic Lateral Sclerosis, vol. 11, no 3, , p. 289-292 (lire en ligne [PDF]).
- A. Chiò, E. Herrero Hernandez, G. Discalzi, P. Ghiglione, N. Di Vito, A. Calvo, M. Vercellino, F. Plano et R. Mutani, « Conjugal amyotrophic lateral sclerosis: suggestion for the implication of environmental factors », Amyotrophic Lateral Sclerosis, vol. 2, no 3, , p. 165-166.
- Brown D. « Lou Gehrig's disease claims Gulf veterans ». Washington Post. August 3, 1999.
- R.D. Horner& al. « Occurrence of amyotrophic lateral sclerosis among Gulf War » (PDF) ; veterans Neurology September 23, 2003 61:742-749.
- Karsarskis EJ, Brooks BR, Ross MA. Persian « Gulf veterans with amyotrophic lateral sderosis (ALS) » : report of a review panel. Washington, DC: Office of the Under-secretary for Health, Department of Veterans Affairs, 1999.
- Homer, R. D. ; Kamins, K. G. ; Feussner, J. R. ; Grambow, S. C. ; Hoff-Lindquist, J.(2002) « Occurrence of Amyotropic Lateral Sclerosis Among Gulf War Veterans » ; Descriptive Note : Rept. for 2 Aug 1990-31 Jul 1991 Corporate Author : Naval Health research center San Diego CA
- Horner RD, Kamins KG, Feussner JR, Grambow SC, Hoff- Lindquist J, Harati Y, et al. « Occurrence of amyotrophic lateral sclerosis among Gulf War veterans ». Neurology. 2003; 61:742-9.
- (en)[PDF] Adriano Chiò, Andrea Calvo, Maurizia Dossena, Paolo Ghiglione, Roberto Mutani, Gabriele Mora, « ALS in Italian professional soccer players: The risk is still present and could be soccer-specific (ALS chez les footballeurs italiens professionnels ; le risque est toujours présent, et pourrait être spécifique aux footballeurs) », Amyotrophic Lateral Sclerosis Jan 2009, 10(4):205-9.
- Henri Haget, « Foot: le syndrome du Calcio », L'Express.fr, (lire en ligne)
- Voir sur lematin.ch.
- A. Chiò, G. Logroscino, O. Hardiman, R. Swingler, D. Mitchell, E. Beghi et B.G. Traynor, « Prognostic factors in ALS: A critical review », Amyotrophic Lateral Sclerosis, vol. 10, nos 5-6, , p. 310-323 (résumé).
- Chiò et al, 2005, revue Brain.
Voir aussi
Liens externes
- Ressources relatives à la santé :
- Orphanet
- (en) Classification internationale des soins primaires
- (en) Diseases Ontology
- (en) DiseasesDB
- (en + es) Genetic and Rare Diseases Information Center
- (en) Héritage mendélien chez l'humain
- (en) Héritage mendélien chez l'humain
- (sv) Internetmedicin
- (en) Medical Subject Headings
- (en + es) MedlinePlus
- (en) NCI Thesaurus
- (no + nn + nb) Store medisinske leksikon
- (cs + sk) WikiSkripta
- « Maladie de Charcot : la plus fréquente des maladies rares », La Méthode scientifique, France Culture, 23 mai 2022.
 Portail de la médecine
Portail de la médecine  Portail des neurosciences
Portail des neurosciences