Chronic multifocal Langerhans cell histiocytosis
| Chronic multifocal Langerhans cell histiocytosis | |
|---|---|
| Other names: Hand–Schüller–Christian disease, multifocal eosinophilic granuloma[1] | |
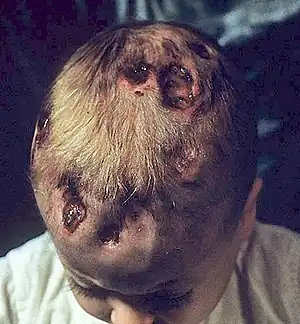 | |
| A child with Hand-Schüller-Christian disease | |
| Specialty | Dermatology |
| Symptoms | Bulging eyes, breakdown of areas of bone, diabetes insipidus[2] |
| Complications | Bone fractures (including spinal fracture), spinal cord injury, loss of teeth[3] |
| Usual onset | 2 to 6 years old[4] |
| Duration | Long term[2] |
| Causes | Genetic mutation during early development[3] |
| Diagnostic method | Tissue biopsy[5][6] |
| Differential diagnosis | Letterer-Siwe disease, mastocytosis[2][3] |
| Treatment | Prednisone, vinblastine, cytarabine, mercaptopurine[3] |
| Prognosis | Variable[6] |
| Frequency | Rare[7] |
Chronic multifocal Langerhans cell histiocytosis, formally known as Hand-Schüller-Christian syndrome, is a type of Langerhans cell histiocytosis (LCH), which can affect multiple organs.[1][3][5] Its three main features are bulging eyes, breakdown of areas of bone (often in the skull), and diabetes insipidus (excessive thirst and dilute urine).[2] However, around 75% of cases do not have all three features.[2] Other features may include an asymmetrical face, ear infections, and advanced gum disease.[2]
It is due to a genetic mutation in the MAPKinase pathway that occurs during early development.[3] The diagnosis may be suspected based on symptoms and MRI and confirmed by tissue biopsy.[5][6] Blood tests may show low red blood cells, and occationally low white blood cells and low platelets.[2]
Treatment may include some combination of prednisone, vinblastine, cytarabine, and mercaptopurine.[3] The outlook depends on how many and how much organs are affected.[6] In some people the condition is life-threatening.[6] The disease is rare.[7] Most cases occur in the 2 to 6 year old age group.[4] Boys are more commonly affected than girls.[2]
It was previously named after the American pediatrician Alfred Hand Jr., the Austrian neuroradiologist Arthur Schüller, and the American pathologist Henry Asbury Christian, who described it in 1893, 1915 and 1919, respectively.[8] Before the Histiocyte Society classified histiocytoses in the 1980s, LCH was known as "histiocytosis X", where "X" denoted the unknown cause at the time.[9][10]
Signs and symptoms
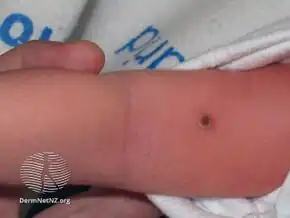
A combination of three features are seen in 25% of people with the condition, which usually presents before the age of five; one or both bulging eyes, breakdown of bone (lytic bone lesions often in the skull in a 'punched out' pattern), and diabetes insipidus (excessive thirst and passing urine).[2][4] Around 75% of cases do not have all three traditional features.[2]
Involvement of the skull is usually accompanied with jaw and basal skull involvement.[11] The face may look asymmetrical and ear infections are common.[2] Between 5 and 75% of cases have been reported to present with signs in the mouth.[2] These include mouth ulcers, bad breath, swollen gums, loose teeth and an unpleasant taste.[2] Destruction of part of the jaw bone may give the appearance of advanced gum disease.[2]
Complications include fractures of long bones, compression of vertebrae, spinal cord damage, swollen and bleeding gums and loss of teeth.[3]
Cause
Chronic multifocal Langerhans cell histiocytosis is due to a genetic mutation in the MAPKinase pathway that occurs during early development.[3] While due to a genetic mutation, it is not inherited from a persons parents.[3]
Some sources such as the National Cancer Institute describe it as a type of cancer,[12] while other sources specifically state it is not a type of cancer.[13]
Diagnostics
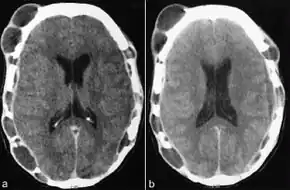
Tests usually include imaging using MRI.[5] Findings include breakdown of bone and thickening of the pituitary stalk. The perivascular space may appear prominent, the pituitary gland cystic and there may be signs in the white matter, a mass in the hypothalamus and enhancement of the meninges.[5]
X-ray findings typically demonstrate sharp "punched out" lesions in the skull. The destruction of alveolar bone is usually more generalised and may appear as displaced teeth.[2]
Blood tests may show anaemia, and less commonly a low white blood cell count and low platelet count.[2] Although the disease was once thought to be a lipid storage disease as the lesions have a high cholesterol content, the blood cholesterol is usually normal.[2][15] Diagnosis is confirmed by tissue biopsy.[5][6]
Epidemiology
The disease is rare.[7] 70% of cases present before the age of 15.[16] Most cases occur in the 2 to 6 year old age group.[4]
Prognosis
The outlook depends on the extent of organ involvement.[6] The prognosis is poor if the disease presents in a young person with many affected organs. In a newborn with skin lesions only, the outlook is better.[6] The prognosis is poor if the liver, spleen, lung, or bone marrow is affected.[6] A good response to chemotherapy within the first six weeks of treatment, has been found to indicate a better prognosis. In some people the condition is life-threatening.[6]
History
MRI and CT scan findings in a mummy have revealed evidence of the disease dating back to 900-790. B.C.[17]
Chronic multifocal Langerhans cell histiocytosis was previously known as Hand–Schüller–Christian disease, and was named for the American pediatrician Alfred Hand Jr.,[8] the Austrian neuroradiologist Arthur Schüller,[8][18] and the American pathologist Henry Asbury Christian,[8][19] who described it in 1893,[8][20] 1915[8][21] and 1919, respectively.[8][22] Shortly afterwards, Letterer in 1924 and Siwe in 1933 described a fatal condition in children who presented with large livers and spleens, large lymph nodes and bone damage. In 1940, Louis Litchtenstein and Henry L. Jaffe described a self-limiting disease characterised by "isolated bone lesions".[16] A common feature of all these conditions was revealed to be the histological findings of large numbers of histiocytes in the tissue biopsies, leading Litchtenstein to propose that the three described conditions were part of a spectrum of a disease he named "Histiocytosis X", where "X" denoted the unknown cause at the time.[16] In 1973, Christian Nezelof recognised the abnormal cell as a 'Langerhans-like' cell, however it took another ten years for the disease to be accepted as one entity and the term 'Langerhans cell histiocytosis' to be internationally recognised. In 1987, the Histiocyte Society published their classification of the histiocyte disorders together with criteria for diagnosis and clinical assessment of Langerhans cell histiocytosis.[9]
References
- 1 2 "Orphanet: OBSOLETE: Hand Schüller Christian disease". www.orpha.net. Archived from the original on 15 November 2017. Retrieved 4 December 2020.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 Rajendran, R. (2009). "17. Diseases of bone and joint (non-neoplastic and non-infectious disorders of bone, skeletal dysplasias/dysostoses, constitutional bone disorders)". In Rajendran, R.; Sivapathasundharan, B. (eds.). Shafer's Textbook of Oral Pathology (6th ed.). Elsevier. p. 744. ISBN 978-81-312-1570-8. Archived from the original on 2021-08-29. Retrieved 2020-12-02.
- 1 2 3 4 5 6 7 8 9 10 "Langerhans Cell Histiocytosis". NORD (National Organization for Rare Disorders). Archived from the original on 10 June 2021. Retrieved 4 December 2020.
- 1 2 3 4 5 Stanway, Amy (2005). "Langerhans cell histiocytosis | DermNet NZ". dermnetnz.org. Archived from the original on 14 August 2021. Retrieved 4 December 2020.
- 1 2 3 4 5 6 "Hand-Schüller-Christian Disease | American Journal of Neuroradiology". www.ajnr.org. Archived from the original on 29 August 2021. Retrieved 2 December 2020.
- 1 2 3 4 5 6 7 8 9 10 "Langerhans cell histiocytosis | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 10 January 2021. Retrieved 5 December 2020. Updated 2017
- 1 2 3 Chang, Karen L.; Snyder, David S. (2007). "17. Langerhans Cell Histiocytosis". In Stephen M. Ansell (ed.). Rare Hematological Malignancies. Springer Science & Business Media. p. 383. ISBN 978-0-387-73743-0. Archived from the original on 2021-08-29. Retrieved 2020-12-04.
- 1 2 3 4 5 6 7 Pluot, Etienne; Davies, Mark; James, Steven L. J. (2009). "Who was who in bone tumours". In Davies, A. Mark; Sundaram, Murali; James, Steven J. (eds.). Imaging of Bone Tumors and Tumor-Like Lesions: Techniques and Applications. Springer. p. 681. ISBN 978-3-540-77982-7. Archived from the original on 2021-08-28. Retrieved 2020-12-02.
- 1 2 Broadbent, V.; Egeler, R. M.; Nesbit, M. E. (September 1994). "Langerhans cell histiocytosis--clinical and epidemiological aspects". The British Journal of Cancer. Supplement. 23: S11–S16. ISSN 0306-9443. PMC 2149699. PMID 8075001. Archived from the original on 2021-08-28. Retrieved 2020-12-04.
- ↑ The Writing Group of the Histiocyte Society (1987). "Histiocytosis syndromes in children. Writing Group of the Histiocyte Society". Lancet. 1 (8526): 208–9. doi:10.1016/S0140-6736(87)90016-X. PMID 2880029. S2CID 54351490.
- ↑ Odell, Edward W (2017). "12. Non-odontogenic tumours of the jaw". Cawson's Essentials of Oral Pathology and Oral Medicine (9th ed.). Elsevier. p. 190. ISBN 978-0-7020-4982-8. Archived from the original on 2021-08-28. Retrieved 2020-12-05.
- ↑ "Langerhans Cell Histiocytosis Treatment (PDQ®)–Patient Version - National Cancer Institute". www.cancer.gov. 16 July 2010. Archived from the original on 29 October 2021. Retrieved 5 December 2020.
- ↑ "Langerhans' cell histiocytosis". www.gosh.nhs.uk. Archived from the original on 24 May 2021. Retrieved 5 December 2020.
- ↑ Cugati, G; Singh, M; Pande, A; Ramamurthi, R; Vasudevan, MC (July 2011). "Hand Schuller Christian disease". Indian journal of medical and paediatric oncology : official journal of Indian Society of Medical & Paediatric Oncology. 32 (3): 183–4. doi:10.4103/0971-5851.92835. PMID 22557792.
- ↑ Schuknecht, Harold F.; Perlman, Henry B. (1 September 1948). "LVI Hand-Schüller-Christian Disease and Eosinophilic Granuloma of the Skull". Annals of Otology, Rhinology & Laryngology. 57 (3): 643–676. doi:10.1177/000348944805700305. ISSN 0003-4894. Archived from the original on 28 August 2021. Retrieved 2 December 2020.
- 1 2 3 C.A.C. Pickering; L. Doyle; K.B. Carroll (1981). "4. Honeycomb lung". Interstitial Lung Disease. MTP Press. p. 83-85. ISBN 978-94-009-8086-0. Archived from the original on 2021-08-28. Retrieved 2020-12-04.
- ↑ Cavka, Mislav; Petaros, Anja; Ivanac, Gordana; Aganović, Lejla; Janković, Ivor; Reiter, Gert; Speier, Peter; Nielles-Vallespin, Sonja; Brkljacić, Boris (March 2012). "A probable case of Hand-Schueller-Christian's disease in an Egyptian mummy revealed by CT and MR investigation of a dry mummy". Collegium Antropologicum. 36 (1): 281–286. ISSN 0350-6134. PMID 22816232. Archived from the original on 2021-08-28. Retrieved 2020-12-04.
- ↑ Schindler, E. (August 1997). "Arthur Schüller: pioneer of neuroradiology". AJNR. American journal of neuroradiology. 18 (7): 1297–1302. ISSN 0195-6108. PMID 9282858. Archived from the original on 2021-08-28. Retrieved 2020-12-02.
- ↑ "Obituary; Henry Asbury Christian". New England Journal of Medicine. 245 (23): 912–913. 6 December 1951. doi:10.1056/NEJM195112062452312. ISSN 0028-4793. PMID 14882449. Archived from the original on 28 August 2021. Retrieved 3 December 2020.
- ↑ A. Hand. Polyuria and tuberculosis. Proceedings of the Pathological Society of Philadelphia, 1893, 16: 282-284. Archives of Pediatrics, New York, 1893: 10: 673-675.
- ↑ Schüller, Arthur (1915). Über eigenartige Schädeldefekte im Jugendalter, Fortschr. a. d. Geb. d. Röentgenstrahlen 23: 12–18.
- ↑ H. Christian. Defects in membranous bones, exophthalmos, and diabetes insipidus; an unusual syndrome of dyspituitarism. In: Contributions to medical and biological research, dedicated to Sir William Osler. New York, P. B. Hoeber, 1919, 1: 390-401. Medical Clinics of North America, Philadelphia, PA., 1920; 3: 849-871.
External links
| Classification |
|---|