Inflammasome
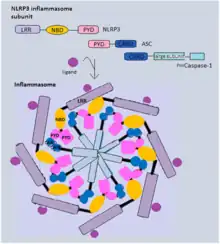
Inflammasomes are cytosolic multiprotein oligomers of the innate immune system responsible for the activation of inflammatory responses.[1][2] Activation and assembly of the inflammasome promotes proteolytic cleavage, maturation and secretion of pro-inflammatory cytokines interleukin 1β (IL-1β) and interleukin 18 (IL-18), as well as cleavage of Gasdermin-D.[2][3] The N-terminal fragment resulting from this cleavage induces a pro-inflammatory form of programmed cell death distinct from apoptosis, referred to as pyroptosis, and is responsible for secretion of the mature cytokines, presumably through the formation of pores in the plasma membrane.[2] In the case of dysregulation of inflammasome activation, an assortment of major diseases, such as cancer, autoimmune, metabolic and neurodegenerative diseases may arise.[2][4]
Traditionally, inflammasomes have mainly been studied in professional immune cells of the innate immune system, such as macrophages. More recent studies, however, indicate high levels of inflammasome component expression in epithelial barrier tissues, where they have been shown to represent an important first line of defense.[5] Inflammasome activation is initiated by different kinds of cytosolic pattern recognition receptors (PRRs) that respond to either microbe-derived pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) generated by the host cell.[5] Pattern recognition receptors involved in inflammasomes comprise NLRs (nucleotide-binding oligomerization domain and leucine-rich repeat-containing receptors) as well as AIM2 (absent in melanoma 2), IFI16 (IFN-inducible protein 16 ) as well as pyrin.[2]
Through their caspase activation and recruitment domain (CARD) or pyrin domain (PYD), the inflammasome receptors interact with the adaptor protein ASC, which then recruits pro-caspase-1 via its CARD domain and activates the effector caspase through proteolytic cleavage.[2] The activated caspase-1 finally cleaves the immature pro-inflammatory cytokines pro-IL-1β and pro-IL-18, as well as Gasdermin-D, which are responsible for inflammatory signaling and pyroptotic cell death, respectively.[2] In addition to these so-called canonical inflammasomes, different studies also described non-canonical inflammasome complexes that act independently of caspase-1. In mice, the non-canonical inflammasome is activated by direct sensing of cytosolic bacterial lipopolysaccharide (LPS) by caspase-11, which subsequently induces pyroptotic cell death.[2] In human cells, the corresponding caspases of the non-canonical inflammasome are caspase 4 and caspase 5.[2]
History
The inflammasome was discovered by the team of Jürg Tschopp, at the University of Lausanne, in 2002.[3][6] Tschopp and team were able to articulate the inflammasome's role in diseases such as gout and type 2 diabetes.[6] They found that a variety of danger signals could provoke a response from an inflammasome including viral DNA, muramyl dipeptide (MDP), asbestos, and silica.[6] Tschopp and his colleagues found a connection between metabolic syndrome and NLRP3, a subset type of inflammasome.[6] Within their research on NLRP3, they were able to show that when NLRP3 is inhibited, an immunosuppressive behavior of type I interferon was exhibited.[6] Ultimately, the work of Tschopp and his team led to the research and eventual treatments of many major diseases and ailments.[6] In 2002, it was first reported by Martinon et al.[3] that a subset of NLRs named NLRP1 were able to assemble and oligomerize into a common structure which collectively activated the caspase-1 cascade, thereby leading to the production of pro-inflammatory cytokines especially IL-1B and IL-18. This NLRP1 multi-molecular complex was dubbed the ‘inflammasome’, which spurred much interest in the following years; since then, several other inflammasomes were discovered, two of which are also NLR subsets—NLRP3 and NLRC4. In 2009, Hornung et al.[7] classified an inflammasome of the PYHIN (pyrin and HIN domain-containing protein) family, termed absent in melanoma 2 (AIM2) which assembles upon sensing foreign cytoplasmic double-stranded DNA (dsDNA). Notably, the pyrin domain of the adaptor protein ASC has recently been shown to function as a prion-like domain, through a self-perpetuating manner upon activation.[8]
Role in innate immunity
During an infection, one of the first forms of defense employed by the innate immune response is a group of pattern recognition receptors (PRRs) encoded in the germline to recognize molecular patterns expressed by invading pathogens (PAMPs). These may either be on the membrane surface e.g. Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) or inside the cytoplasm e.g. Nod-like receptors (NLRs) and RIG-I-like receptors (RLRs). Inflammasome complexes are activated by a subset of cytosolic PRRs that recognize a variety of microbe-derived PAMPs and DAMPs generated by the cell itself.[5] As part of the innate immune system, inflammasomes play an important role in the induction of inflammatory cascades and coordination of host defenses, both via the activation and secretion of pro-inflammatory cytokines and the induction of a specialized form of immune-stimulatory programmed cell death termed pyroptosis.[2] Traditionally, inflammasomes have mainly been studied in professional innate immune cells such as macrophages. More recently, however, several studies described various epithelial inflammasomes and highlighted their crucial role as a first line of defense.[5][9] Since epithelial cells line important barrier tissues such as the intestines, their inflammasomes are optimally positioned to recognize invading microbes at the first point of interaction.
Inflammatory cascade
Analogous to the apoptosome, which activates apoptotic cascades, the inflammasome activates a pyroptotic inflammatory cascade.[10] Once active, the inflammasome binds to pro-caspase-1 (the precursor molecule of caspase-1), either homotypically via its own caspase activation and recruitment domain (CARD) or via the CARD of the adaptor protein ASC which it binds to during inflammasome formation. In its full form, the inflammasome appositions together many p45 pro-caspase-1 molecules, inducing their autocatalytic cleavage into p20 and p10 subunits.[11] Caspase-1 then assembles into its active form consisting of two heterodimers with a p20 and p10 subunit each. Once active, it can then carry out a variety of processes in response to the initial inflammatory signal. These include the proteolytic cleavage of pro-IL-1β at Asp116 into IL-1β,[3] cleavage of pro-IL-18 into IL-18, as well as cleavage of Gasdermin-D to release its N-terminal fragment responsible for the induction of pyroptosis, an immunostimulatory form of programmed cell death in which the cell releases its cytoplasmic content to induce pro-inflammatory signaling.[2][12] IL-1β and IL-18 released following inflammasome activation were found to induce IFN-γ secretion and natural killer cell activation,[13] cleavage and inactivation of IL-33,[14] DNA fragmentation and cell pore formation,[15] inhibition of glycolytic enzymes,[16] activation of lipid biosynthesis[17] and secretion of tissue-repair mediators such as pro-IL-1α.[18] Additionally, AIM2 contains a HIN200 domain which senses and binds foreign cytoplasmic dsDNA[19] and activates NF-κB,[7] a role that is crucial in bacterial and viral infection.
NLR-subset inflammasomes
NLRP1, NLRP3, NLRP6 and NLRC4 are subsets of the NLR family and thus have two common features: the first is a nucleotide-binding oligomerization domain (NOD) which is bound by ribonucleotide-phosphates (rNTP) and is important for self-oligomerization.[20] The second is a C-terminal leucine-rich repeat (LRR), which serves as a ligand-recognition domain for other receptors (e.g. TLR) or microbial ligands. NLRP1 has been found in neurons, while both NLRP3 and NLRC4 (IPAF) have been identified in microglial cells.[21]
NLRP1
- See NLRP1 for gene information
Structure
In addition to NOD and LRR, NLRP1 contains at its N-terminal a pyrin domain (PYD) and at its C-terminus an FIIND motif and a CARD, which distinguishes it from the other inflammasomes. While there is only one NLRP1 protein present in humans, rodents were found to have different paralogues.[2] Upon activation, the C-terminal CARD homotypically interacts with the CARD of procaspase-1 or procaspase-5, while its N-terminal PYD homotypically interacts with the PYD of adaptor protein ASC, whose CARD can then recruit another pro-caspase-1. The overall recruitment and cleavage of procaspase-1 can then activate all downstream caspase-1 pathways.
Activation
NLRP1B in mice and NLRP2 in rats were found to be responsive to Bacillus anthracis lethal toxin.[2] The B. anthracis lethal factor proteolytically cleaves NLRP1B, which leads to ubiquitination of the receptor and targeting for degradation by the proteasome. This degradation generates a clipped C-terminal fragment, that subsequently binds to the rest of the protein in a non-covalent manner. During this process, a CARD on the C-terminal fragment becomes accessible for inflammasome assembly.[5] So far, this activation mechanism depending on degradation by the proteasome machinery is unique among inflammasomes.[5]
NLRP1 activity is regulated by anti-apoptotic proteins Bcl-2 and Bcl-x(L) which, in resting cells, associate with and inhibit NLRP1 activity.[22]
NLRP3
- See NALP3 for gene information
Structure
In addition to the NOD and LRR domains, NLRP3 contains a PYD domain like NLRP1 and thus activates caspase-1 the same way, using its PYD to recruit ASC. It forms only one oligomer per cell, and its oligomer is made of seven NLRP3 molecules. It is known to be the biggest inflammasome of all, covering about 2 um in diameter.[23]
Activation
NLRP3 oligomerization is activated by a large number of stimuli, including both PAMPs and DAMPs, which has implicated studies into its activation pathway. Examples of such stimuli from the DAMP group are crystalline matter such as monosodium urate (MSU) crystals, alum or asbestos, calcium influx, mitochondrial reactive oxygen species (ROS), and extracellular ATP.[2][24] The NLRP3 inflammasome was also found to respond to PAMPs of different pathogens, such as viruses, e.g. influenza A,[25] bacteria, e.g. Neisseria gonorrhoeae,[26] and bacterial toxins e.g. nigericin and maitotoxin.[1] The downstream point where all the different NLRP3 stimuli converge was found to below intracellular potassium concentrations.[2] NLRP3 inflammasome activation by cholesterol crystals and MSU crystals increase NLRP3-induced IL-1β-production[27][28] and this process is thought to be abrogated in atherosclerosis and gout, where these crystals form in the cell. It has also been proven that inorganic particles like titanium dioxide, silicon dioxide and asbestos can trigger activation of the inflammasome.[29] One study even indicates that NLRP3 inflammasome activation is involved in sleep regulation.[30] Recent studies show that NLRP3 inflammasome-mediated neuroinflammation is involved in secondary brain injury after intracerebral hemorrhage.[31]
Inhibition
There has been some progress in the development of NLRP3 inhibitors some include: Dapansutrile, and diarylsulfonylurea MCC-950.[32][33]
NAIP/NLRC4
- See NLRC4 for gene information
Structure
NLRC4 (also known as IPAF) is the only known subset of the NLRC family to form an inflammasome and contains only a CARD domain in addition to the NOD and LRR, which it uses to recruit the adaptor protein ASC or pro-caspase-1 directly. In contrast to other inflammasomes, NLRC4 applies NAIPs (NLR family apoptosis inhibitory proteins) as upstream receptors.[2]
Activation
The NAIP/NLRC4 inflammasome is involved in host defense.[34] NAIPs are activated by binding to the bacterial PAMPs in the cytosol, given by the rod (NAIP2) and needle (NAIP1) components of the bacterial type-3 secretion system (T3SS), as well as flagellin, the molecular building block of flagella (NAIP5 and 6).[2][35] Following ligand binding, NAIPs interact with NLRC4 to initiate the assembly of the NAIP/NLRC4 inflammasome, which then recruits and activates pro-caspase-1 via its CARD domain.[35]
Palmitate has been shown experimentally to induce the NLRC4 inflammasome without any bacteria present. This may give insight to other functions the inflammasome may have in the immune system, and also suggests that the inflammasome can respond to more than just bacteria. The NLRC4 inflammasome is regulated by cyclic adenosine monophosphate (cAMP).[34]
The Adaptor ASC
Apoptosis-associated speck like protein containing a caspase recruitment domain (ASC or Pycard) plays a key role in activation of the inflammasome.[4] ASC helps recruit caspase-1 to associate with NLRs in the inflammasome complex via its CARD domain.[36]
ASC also has duties independent of the inflammasome as it has been shown to be required for MHC class II to present antigenic peptides in dendritic cells.[4]
AIM2 and IFI16
AIM2
Main article: AIM2
AIM2 is an acronym for absent in melanoma 2, and is sometimes also referred to as Interferon-inducible protein.
Structure
AIM2 is a 343 amino acid non-NLR family protein with pyrin (DAPIN) and a HIN-200 domains,[37] the former of which is activated in AIM2 by dsDNA.[38]
Function
The AIM2 inflammasome is a detector of cytosolic double stranded DNA (dsDNA) and plays an important role in the coordination of immune defense to DNA virus infections, as well as intracellular bacterial infections.[2] AIM2 is activated by viral dsDNA, bacterial dsDNA and also aberrant host dsDNA.,[39][40] and consequently it has been linked to different human diseases. For example autoinflammation in psoriasis disease was found to be connected to recognition of self-DNA by AIM2.[2] Furthermore, activation of the AIM2 is supposed to play role in autoimmune responses during the autoimmune disease systematic lupus erythematosus. The AIM2 inflammasome is also activated by pharmacological disruption of nuclear envelope integrity.[41] AIM2 binds dsDNA with its C-terminal HIN-200 domain.[42][43][39] The PYDdomain of AIM2 homotypically interacts by PYD-PYD interactions with ASC. The ASC CARD domain recruits pro-caspase-1 into the complex. Caspase-1 activates maturation of proinflammatory cytokines (IL-1β, IL-18).
IFI16
Like AIM2, IFI16 (IFN-inducible protein 16) belongs to the PYHIN (pyrin and HIN domain-containing) family. IFI16 in humans, and IFI204, the mouse orthologue play an important role in regulating the production of IFN during both bacterial and viral infections.[2] In contrast to AIM2, IFI16 is a nuclear DNA sensor.[5] Following interaction with viral DNAs, IFI16 was shown to recruit caspase-1 through interaction with ASC, resulting in cell death of CD4+ T cells in response to HIV infection.[2]
Pyrin
Pyrin is encoded by the gene MEFV and is sometimes also referred to as TRIM20 or marenostrin.[2]
Structure
Pyrin is composed of a PYD domain, two B-boxes, a coiled-coil domain as well as a B30.2 domain at its C-terminus, whereby the latter is only present in human pyrin.[2]
Function
Assembly of the pyrin inflammasome is triggered by bacterial toxins as well as effector proteins via the detection of pathogen-dirven disturbances in cytoskeleton dynamics.[2] More specifically, pyrin detects inactivation of the Rho GTPase RHOA by these bacterial factors.[2] Following detection of RHOA inactivation, pyrin interacts with ASC through its N-terminal PYD domain to induce activation of caspase-1.[2]
Non-canonical inflammasomes
The name non-canonical inflammasomes has its origin in the fact that these inflammasome complexes are independent of caspase-1. In mice, the non-canonical inflammasome is dependent on caspase-11 whereas human non-canonical inflammasomes rely on caspase 4 and caspase 5.[2] All of these caspases are able to directly bind intracellular LPS and subsequently form macromolecular complexes mediating Gasdermin-D cleavage and induction of pyroptotic cell death.[5] In addition, non-canonical inflammasomes may also indirectly activate the NLRP3 inflammasome by triggering potassium efflux through membrane pores formed by Gasdermin-D.[5] The NLRP3 inflammasome can then mediate processing of pro-inflammatory cytokines and result in release of IL-1β and IL-18 in response to non-canonical inflammasome activation.[2]
Epithelial inflammasomes
Traditionally, inflammasomes have mainly been studied in professional cells of the innate immune system such as macrophages. More recently, however, inflammasome-mediated defense was found to play a crucial role as a first line of defense against bacterial onslaught also at a variety of epithelial sites.[5][9][44] Considering the importance of epithelia as common pathogen entry sites as a result of their location at the interface to the environment, it seems intuitive that epithelial tissues do not merely fulfill a function as a physical barrier, but additionally initiate a defensive response upon initial contact with the pathogen. In agreement with this, different inflammasome components were found to be expressed in a variety of epithelial tissues.[5][9] Expression of innate immune components at epithelial barriers furthermore facilitates pathogen detection given that expression of virulence factors and hence exposure of PAMPs is required for the breaching of these barriers during invasion, whereas these factors might be downregulated when the pathogen interacts with professional immune cells at later stages of infection.[44] Epithelial inflammasomes have mainly been studied in the intestinal mucosa, but there is also evidence for inflammasomes in other types of epithelial such as the urinary bladder epithelium.[44]
Epithelial NAIP/NLRC4
The NAIP/NLRC4 inflammasome is the best described epithelial inflammasome and plays an important role in the restriction of intraepithelial bacterial populations during early stages of enterobacterial infection with e.g. Salmonella and Citrobacter rodentium.[9][44] Intracellular bacteria trigger activation of the inflammasome, which results in specific expulsion of infected epithelial cells from the epithelium in order to reduce the bacterial loads.[9][44] This process is also referred to as epithelial cell extrusion, and occurs without compromising the integrity of the epithelial barrier. Furthermore, the NAIP/NLRC4 inflammasome was found to reduce tumor loads in a mouse model of colorectal carcinoma (CRC), by triggering the removal of tumor-initiating cells.[9]
Epithelial non-canonical inflammasomes
While the murine caspase-11 is mainly expressed in macrophages, human caspase-4 is also expressed at high levels in intestinal epithelial cells.[9] Similar to what was observed for the epithelial NAIP/NLRC4 inflammasome, human epithelial cells were shown to undergo caspase-4-dependent, caspase-1-independent cell death and extrusion in response to infection with the enteropathogens such as Salmonella, Shigella flexneri or Escherichia coli.[9] Furthermore, secretion of IL-18 could be triggered by cytosolic LPS in epithelial cells.[9]
Consequences of epithelial inflammasome activation
Activation of epithelial inflammasomes in response to invading pathogens has important cell-autonomous effects on the infected cell itself as well as on its communication with other cell types on a local and global level.[44] These downstream consequences of inflammasome activation can be divided into three categories, namely (1) death of the epithelial cell itself, (2) release of soluble pro-inflammatory molecules, and (3) effector cell recruitment and activation.[44] In addition, epithelial inflammasome activation induces contraction of epithelial layers[45] and prevents integrity loss at later stages of infection.[46]
Epithelial cell death
In order to maintain the integrity of the epithelial barrier, cell death and subsequent extrusion of the infected cell have to occur in a coordinated manner to ensure sealing of the gap in the epithelium by neighboring cells.[44] Epithelial cell death can be triggered in a direct, cell-autonomous manner by inflammasome activation itself, as well as by local recruitment of other death-inducing cell types, or global inflammation, resulting in increased epithelial turnover that removes both infected and uninfected cells.[44] The most important consequence of epithelial cell death is the reduction of epithelial pathogen loads in order to maintain barrier integrity.[44]
Release of soluble immune mediators
Inflammasome activation is known to trigger cleavage, activation and secretion of pro-inflammatory IL-1β and IL-18,[2] which then recruit different types of effector cells and coordinate the innate immune response.[44] Expression of pro-IL-1β is dependent on Toll-like-receptor signaling, and hence intestinal epithelial cells produce very low levels of IL-1β themselves. Pro-IL-18, on the other hand, is constitutively expressed by different kinds of epithelial cells, and readily secreted upon inflammasome activation.[44] The IL-18 secreted by the epithelium can induce production of IFN-γ by different cell types.
Recruitment and activation of effector cells
Importantly, inflammasome-derived IL-18 is also involved in recruitment of natural killer (NK) cells, that play a crucial role at early stages of innate immune responses.[44] Furthermore, IL-18 can also stimulate the effector functions of NK cells that accumulated at the site of infection.[44] The activated NK cells can then help in restricting the pathogen loads and spread to systemic sites at later time points compared to epithelium-autonomous induction of cell death.[44] Finally, NK cells also secrete IFN-γ in order to recruit other inflammatory cell types.[44]
In a study of UPEC infection of the urinary bladder epithelium, epithelial cells were found to secrete IL-1β at high levels in response to bacterial infection. The study reported IL-1β secretion to be dependent on the NLRP3 inflammasome and caspase-1, and the secreted pro-inflammatory cytokine was required for recruitment of mast cells to the site of infection. The mast cells then induce a lytic form of cell death in the epithelium by secreting granules that are taken up by the epithelium.[44]
Besides NK cells and mast cells, neutrophils are other important innate immune effector cells that infiltrate the infected tissue after breaching of epithelial barriers by pathogens. Both IL-1β and IL-18 secreted in response to inflammasome activation are involved in the recruitment of neutrophils.[44] Once they have reached the infected tissue, the neutrophils assist in immobilizing and eliminating the invading pathogens, whereby they either directly engulf or kill invading microbes.[44] Furthermore, they secrete inflammatory mediators such as IFN-γ and IL-22. IFN-γ is known to drive activation of microbicidal capacity of mononuclear phagocytes. IL-22, on the other hand, will strengthen the epithelial barrier.[44] Finally, neutrophils are responsible for the elimination of bacteria that are trapped within pyroptotic macrophages.[44]
Dysregulated inflammasome activity
Problems with regulating inflammasomes have been linked to several autoimmune diseases such as type I and type II diabetes, inflammatory bowel disease (IBD), gouty arthritis, multiple sclerosis, and vitiligo as well as auto-inflammatory disorders.[4][47] These diseases and disorders have been connected to too much or too little secretion of the pro-inflammatory cytokines that the inflammasome is responsible for. Gain-of-function mutations in inflammasome components are also known to cause Cryopyrin-associated periodic syndrome (CAPS), a group of congenital diseases characterised by IL-1β-mediated systemic inflammation.
References
- 1 2 Mariathasan S, Newton K, Monack D, Vucic D, French D, Lee W, Roose-Girma M, Erickson S, Dixit V (2004). "Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf". Nature. 430 (6996): 213–8. Bibcode:2004Natur.430..213M. doi:10.1038/nature02664. PMID 15190255. S2CID 4317409.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 Broz, Petr; Dixit, Vishva M. (July 2016). "Inflammasomes: mechanism of assembly, regulation and signalling". Nature Reviews Immunology. 16 (7): 407–420. doi:10.1038/nri.2016.58. ISSN 1474-1733. PMID 27291964. S2CID 32414010.
- 1 2 3 4 Martinon F, Burns K, Tschopp J (2002). "The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta". Mol Cell. 10 (2): 417–26. doi:10.1016/S1097-2765(02)00599-3. PMID 12191486.
- 1 2 3 4 Ippagunta, Sirish K; Malireddi, R K Subbarao; Shaw, Patrick J; Neale, Geoffrey A; Walle, Lieselotte Vande; Green, Douglas R; Fukui, Yoshinori; Lamkanfi, Mohamed; Kanneganti, Thirumala-Devi (October 2011). "The inflammasome adaptor ASC regulates the function of adaptive immune cells by controlling Dock2-mediated Rac activation and actin polymerization". Nature Immunology. 12 (10): 1010–1016. doi:10.1038/ni.2095. ISSN 1529-2916. PMC 3178750. PMID 21892172.
- 1 2 3 4 5 6 7 8 9 10 11 Winsor, Nathaniel; Krustev, Christian; Bruce, Jessica; Philpott, Dana J.; Girardin, Stephen E. (2019-07-22). "Canonical and noncanonical inflammasomes in intestinal epithelial cells". Cellular Microbiology. 21 (11): e13079. doi:10.1111/cmi.13079. ISSN 1462-5814. PMID 31265745. S2CID 195786609.
- 1 2 3 4 5 6 Dagenais M, Skeldon A, Saleh M (January 2012). "The inflammasome: in memory of Dr. Jurg Tschopp". Cell Death Differ. 19 (1): 5–12. doi:10.1038/cdd.2011.159. ISSN 1476-5403. PMC 3252823. PMID 22075986.
- 1 2 Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA (2009). "AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC". Nature. 458 (7237): 514–8. Bibcode:2009Natur.458..514H. doi:10.1038/nature07725. PMC 2726264. PMID 19158675.
- ↑ Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, Chen ZJ (2014). "Prion-like Polymerization Underlies Signal Transduction in Antiviral Immune Defense and Inflammasome Activation". Cell. 156 (6): 1207–22. doi:10.1016/j.cell.2014.01.063. PMC 4034535. PMID 24630723.
- 1 2 3 4 5 6 7 8 9 Sellin, Mikael E.; Maslowski, Kendle M.; Maloy, Kevin J.; Hardt, Wolf-Dietrich (2015-08-01). "Inflammasomes of the intestinal epithelium". Trends in Immunology. 36 (8): 442–450. doi:10.1016/j.it.2015.06.002. ISSN 1471-4906. PMID 26166583.
- ↑ Bergsbaken, Tessa; Fink, Susan L.; Cookson, Brad T. (February 2009). "Pyroptosis: host cell death and inflammation". Nature Reviews Microbiology. 7 (2): 99–109. doi:10.1038/nrmicro2070. ISSN 1740-1526. PMC 2910423. PMID 19148178.
- ↑ Yamin TT, Ayala JM, Miller DK (1996). "Activation of the native 45-kDa precursor form of interleukin-1 converting enzyme". J Biol Chem. 271 (22): 13273–13282. doi:10.1074/jbc.271.22.13273. PMID 8662843.
- ↑ Xia, Shiyu; Zhang, Zhibin; Magupalli, Venkat Giri; Pablo, Juan Lorenzo; Dong, Ying; Vora, Setu M.; Wang, Longfei; Fu, Tian-Min; Jacobson, Matthew P.; Greka, Anna; Lieberman, Judy (2021-04-21). "Gasdermin D pore structure reveals preferential release of mature interleukin-1". Nature. 593 (7860): 607–611. Bibcode:2021Natur.593..607X. doi:10.1038/s41586-021-03478-3. ISSN 0028-0836. PMID 33883744. S2CID 233351704.
- ↑ Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, Hayashi N, Higashino K, Okamura H, Nakanishi K, Kurimoto M, Tanimoto T, Flavell RA, Sato V, Harding MW, Livingston DJ, Su MS (1997). "Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme". Science. 275 (5297): 206–209. doi:10.1126/science.275.5297.206. PMID 8999548. S2CID 85955985.
- ↑ Cayrol C, Girard JP (2009). "The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1". Proc Natl Acad Sci U S A. 106 (22): 9021–6. Bibcode:2009PNAS..106.9021C. doi:10.1073/pnas.0812690106. PMC 2690027. PMID 19439663.
- ↑ Fink SL, Cookson BT (2006). "Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages". Cellular Microbiology. 8 (11): 1812–25. doi:10.1111/j.1462-5822.2006.00751.x. PMID 16824040. S2CID 23338786.
- ↑ Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M (2007). "The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock". J Biol Chem. 282 (50): 36321–9. doi:10.1074/jbc.M708182200. PMID 17959595.
- ↑ Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG (2006). "Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival". Cell. 126 (6): 1135–45. doi:10.1016/j.cell.2006.07.033. PMID 16990137. S2CID 616037.
- ↑ Keller M, Rüegg A, Werner S, Beer HD (2008). "Active caspase-1 is a regulator of unconventional protein secretion". Cell. 132 (5): 818–31. doi:10.1016/j.cell.2007.12.040. PMID 18329368. S2CID 17330211.
- ↑ Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES (2009). "AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA". Nature. 458 (7237): 509–13. Bibcode:2009Natur.458..509F. doi:10.1038/nature07710. PMC 2862225. PMID 19158676.
- ↑ Ye Z, Lich JD, Moore CB, Duncan JA, Williams KL, Ting JP (2008). "ATP binding by monarch-1/NLRP12 is critical for its inhibitory function". Mol Cell Biol. 28 (5): 1841–50. doi:10.1128/MCB.01468-07. PMC 2258772. PMID 18160710.
- ↑ Jamilloux, Yvan; Pierini, Roberto; Querenet, Mathieu; Juruj, Carole; Fauchais, Anne-Laure; Jauberteau, Marie-Odile; Jarraud, Sophie; Lina, Gérard; Etienne, Jérôme (2013-04-01). "Inflammasome activation restricts Legionella pneumophila replication in primary microglial cells through flagellin detection". Glia. 61 (4): 539–549. doi:10.1002/glia.22454. ISSN 1098-1136. PMID 23355222. S2CID 28910610.
- ↑ Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, Matsuzawa S, Terskikh AV, Faustin B, Reed JC (2007). "Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1". Cell. 129 (1): 45–56. doi:10.1016/j.cell.2007.01.045. PMID 17418785. S2CID 18347164.
- ↑ Stutz A, Golenbock DT, Latz E (2009). "Inflammasomes: too big to miss". J Clin Invest. 119 (12): 3502–3511. doi:10.1172/JCI40599. PMC 2786809. PMID 19955661.
- ↑ Crowley, Shauna M.; Knodler, Leigh A.; Vallance, Bruce A. (2016), Backert, Steffen (ed.), "Salmonella and the Inflammasome: Battle for Intracellular Dominance", Inflammasome Signaling and Bacterial Infections, Springer International Publishing, vol. 397, pp. 43–67, doi:10.1007/978-3-319-41171-2_3, ISBN 9783319411705, PMID 27460804
- ↑ Thomas PG, Dash P, Aldridge JR Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, Kanneganti TD (2009). "The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1". Immunity. 30 (4): 566–75. doi:10.1016/j.immuni.2009.02.006. PMC 2765464. PMID 19362023.
- ↑ Duncan JA, Gao X, Huang MT, O'Connor BP, Thomas CE, Willingham SB, Bergstralh DT, Jarvis GA, Sparling PF, Ting JP (2009). "Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome". Journal of Immunology. 182 (10): 6460–9. doi:10.4049/jimmunol.0802696. PMC 2722440. PMID 19414800.
- ↑ Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J (2006). "Gout-associated uric acid crystals activate the NALP3 inflammasome". Nature. 440 (7081): 237–41. Bibcode:2006Natur.440..237M. doi:10.1038/nature04516. PMID 16407889.
- ↑ Jamilloux, Y; Sève, P; Henry, T (November 2014). "[Inflammasomes in human diseases]". La Revue de Médecine Interne. 35 (11): 730–41. doi:10.1016/j.revmed.2014.04.017. PMID 24907108.
- ↑ Yazdi AS, Guarda G, Riteau N, Drexler SK, Tardivel A, Couillin I, Tschopp J (Nov 2010). "Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1α and IL-1β". Proc Natl Acad Sci U S A. 107 (45): 19449–19454. Bibcode:2010PNAS..10719449Y. doi:10.1073/pnas.1008155107. PMC 2984140. PMID 20974980.
- ↑ Zielinski MR, Gerashchenko D, Karpova SA, Konanki V, McCarley RW, Sutterwala FS, Strecker RE, Basheer R (2017). "The NLRP3 inflammasome modulates sleep and NREM sleep delta power induced by spontaneous wakefulness, sleep deprivation and lipopolysaccharide". Brain Behav Immun. 62: 137–150. doi:10.1016/j.bbi.2017.01.012. PMC 5373953. PMID 28109896.
- ↑ Ren H, Han R, Chen X, Liu X, Wan J, Wang L, Yang X, Wang J (May 2020). "Potential therapeutic targets for intracerebral hemorrhage-associated inflammation: An update". J Cereb Blood Flow Metab. 40 (9): 1752–1768. doi:10.1177/0271678X20923551. PMC 7446569. PMID 32423330. S2CID 218689863.
- ↑ Alzheimer's Drug Discovery Foundation, Dapansutrile. ". "Dapansutrile" (PDF).
- ↑ Coll, Rebecca C.; Robertson, Avril A. B.; Chae, Jae Jin; Higgins, Sarah C.; Muñoz-Planillo, Raúl; Inserra, Marco C.; Vetter, Irina; Dungan, Lara S.; Monks, Brian G.; Stutz, Andrea; Croker, Daniel E. (March 2015). "A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases". Nature Medicine. 21 (3): 248–255. doi:10.1038/nm.3806. ISSN 1546-170X. PMC 4392179. PMID 25686105.
- 1 2 Liu, Li; Chan, Christina (2014). "IPAF inflammasome is involved in interleukin-1β production from astrocytes, induced by palmitate; implications for Alzheimer's Disease". Neurobiology of Aging. 35 (2): 309–321. doi:10.1016/j.neurobiolaging.2013.08.016. PMC 3832124. PMID 24054992.
- 1 2 Zhao, Yue; Shao, Feng (May 2015). "The NAIP-NLRC4 inflammasome in innate immune detection of bacterial flagellin and type III secretion apparatus". Immunological Reviews. 265 (1): 85–102. doi:10.1111/imr.12293. PMID 25879286. S2CID 24423221.
- ↑ Kanneganti, Thirumala-Devi; Lamkanfi, Mohamed; Núñez, Gabriel (2007). "Intracellular NOD-like Receptors in Host Defense and Disease". Immunity. 27 (4): 549–559. doi:10.1016/j.immuni.2007.10.002. PMID 17967410.
- ↑ Anon. (2015). "Entrez Gene: AIM2 absent in melanoma 2 [ Homo sapiens (human)], Gene ID: 9447 (updated on 8-Nov-2015)". Bethesda, MD, USA: National Center for Biotechnology Information, National Library of Medicine. Retrieved 13 November 2015.
- ↑ Lu A, Wu H (2015). "Structural mechanisms of inflammasome assembly". review. FEBS J. 282 (3, February): 435–444. doi:10.1111/febs.13133. PMC 6400279. PMID 25354325.
- 1 2 Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, Hume DA, Stacey KJ (February 2009). "HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA". Science. 323 (5917): 1057–60. Bibcode:2009Sci...323.1057R. doi:10.1126/science.1169841. PMID 19131592. S2CID 43712804.
- ↑ Muruve DA, Pétrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J (March 2008). "The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response". Nature. 452 (7183): 103–7. Bibcode:2008Natur.452..103M. doi:10.1038/nature06664. PMID 18288107. S2CID 205212334.
- ↑ Di Micco, A; Frera, G; Lugrin, J; Jamilloux, Y; Hsu, ET; Tardivel, A; De Gassart, A; Zaffalon, L; Bujisic, B; Siegert, S; Quadroni, M; Broz, P; Henry, T; Hrycyna, CA; Martinon, F (9 August 2016). "AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity". Proceedings of the National Academy of Sciences of the United States of America. 113 (32): E4671–80. doi:10.1073/pnas.1602419113. PMC 4987819. PMID 27462105.
- ↑ Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G (March 2009). "An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome". Nat. Immunol. 10 (3): 266–72. doi:10.1038/ni.1702. PMID 19158679. S2CID 5597950.
- ↑ Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES (March 2009). "AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA". Nature. 458 (7237): 509–13. Bibcode:2009Natur.458..509F. doi:10.1038/nature07710. PMC 2862225. PMID 19158676.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 Sellin, Mikael E.; Müller, Anna A.; Hardt, Wolf-Dietrich (2018-01-19). "Consequences of Epithelial Inflammasome Activation by Bacterial Pathogens". Journal of Molecular Biology. Mechanisms of Inflammasome Activation. 430 (2): 193–206. doi:10.1016/j.jmb.2017.03.031. ISSN 0022-2836. PMID 28454742.
- ↑ Samperio Ventayol, Pilar; Geiser, Petra; Di Martino, Maria Letizia; Florbrant, Alexandra; Fattinger, Stefan A.; Walder, Naemi; Sima, Eduardo; Shao, Feng; Gekara, Nelson O.; Sundbom, Magnus; Hardt, Wolf-Dietrich (2021-04-20). "Bacterial detection by NAIP/NLRC4 elicits prompt contractions of intestinal epithelial cell layers". Proceedings of the National Academy of Sciences. 118 (16): e2013963118. doi:10.1073/pnas.2013963118. ISSN 0027-8424. PMC 8072224. PMID 33846244.
- ↑ Fattinger, Stefan A.; Geiser, Petra; Samperio Ventayol, Pilar; Di Martino, Maria Letizia; Furter, Markus; Felmy, Boas; Bakkeren, Erik; Hausmann, Annika; Barthel-Scherrer, Manja; Gül, Ersin; Hardt, Wolf-Dietrich (May 2021). "Epithelium-autonomous NAIP/NLRC4 prevents TNF-driven inflammatory destruction of the gut epithelial barrier in Salmonella-infected mice". Mucosal Immunology. 14 (3): 615–629. doi:10.1038/s41385-021-00381-y. ISSN 1933-0219. PMC 8075861. PMID 33731826.
- ↑ So, Alexander; Busso, Nathalie (2014). "The concept of the inflammasome and its rheumatologic implications". Joint Bone Spine. 81 (5): 398–402. doi:10.1016/j.jbspin.2014.02.009. PMID 24703401.
Further reading
- Jin T, Xiao TS (February 2015). "Activation and assembly of the inflammasomes through conserved protein domain families". Apoptosis. 20 (2): 151–6. doi:10.1007/s10495-014-1053-5. PMC 4364414. PMID 25398536.
- Lu A, Wu H (February 2015). "Structural mechanisms of inflammasome assembly". review. The FEBS Journal. 282 (3): 435–44. doi:10.1111/febs.13133. PMC 6400279. PMID 25354325.
- Shaw N, Liu ZJ (January 2014). "Role of the HIN domain in regulation of innate immune responses". Molecular and Cellular Biology. 34 (1): 2–15. doi:10.1128/MCB.00857-13. PMC 3911281. PMID 24164899.
- Walsh JG, Muruve DA, Power C (February 2014). "Inflammasomes in the CNS". review. Nature Reviews. Neuroscience. 15 (2): 84–97. doi:10.1038/nrn3638. PMID 24399084. S2CID 7592807.
- Schroder K, Tschopp J (March 2010). "The inflammasomes". review. Cell. 140 (6): 821–32. doi:10.1016/j.cell.2010.01.040. PMID 20303873. S2CID 16916572.
- Jha S, Ting JP (December 2009). "Inflammasome-associated nucleotide-binding domain, leucine-rich repeat proteins and inflammatory diseases". Journal of Immunology. 183 (12): 7623–9. doi:10.4049/jimmunol.0902425. PMC 3666034. PMID 20007570.
- Stutz A, Golenbock DT, Latz E (December 2009). "Inflammasomes: too big to miss". The Journal of Clinical Investigation. 119 (12): 3502–11. doi:10.1172/JCI40599. PMC 2786809. PMID 19955661.
- Fink SL, Cookson BT (April 2005). "Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells". Infection and Immunity. 73 (4): 1907–16. doi:10.1128/IAI.73.4.1907-1916.2005. PMC 1087413. PMID 15784530.
- Martinon F, Tschopp J (August 2005). "NLRs join TLRs as innate sensors of pathogens". review. Trends in Immunology. 26 (8): 447–54. doi:10.1016/j.it.2005.06.004. PMID 15967716.