تضخم الغدة الكظرية الخلقي الشحمي
تضخم الغدة الكظرية الخلقي الشحمي هو اضطراب في الغدد الصماء وهو شكل غير شائع ويمكن أن يكون مميتًا من تضخم الغدة الكظرية الخلقي (CAH). ينشأ من عيوب في المراحل الأولى من إنتاج هرمون الستيرويد: نقل الكوليسترول إلى الميتوكوندريا وتحويل الكوليسترول إلى البريغنينولون - الخطوة الأولى في تخليق جميع هرمونات الستيرويد. يسبب تضخم الغدة الكظرية الخلقي الشحمي نقص القشرانيات المعدنية عند الرضع والأطفال المصابين. الأطفال الذكور يعانون من نقص شديد في التذكير مما يجعل أعضائهم التناسلية الخارجية تبدو أنثوية. الغدد الكظرية كبيرة ومليئة بالكريات الدهنية المشتقة من الكوليسترول.

| تضخم الغدة الكظرية الخلقي الشحمي | |
|---|---|
| معلومات عامة | |
| من أنواع | فرط تنسج الكظرية الخلقي |

تقديم
يمكن تقسيم المشاكل التي تظهر في الأشخاص المصابين بتضخم الغدة الكظرية الخلقي الشحمي إلى:
- نقص القشرانيات المعدنية.
- نقص الكورتيكويد السكري (الجلوكوكورتيكويد).
- نقص الستيرويد الجنسي.
- الأضرار التي لحقت الغدد التناسلية الناجمة عن تراكم الدهون.
نقص القشرانيات المعدنية
معظم الأطفال الذين يولدون مصابين بتضخم الغدة الكظرية الخلقي الشحمي لديهم أعضاء تناسلية أنثوية بدرجة كافية بحيث لا يشتبه في أي مرض عند الولادة. نظرًا لأن منطقة الكبيبات الكظرية غير متمايزة وغير نشطة قبل الولادة ، فإنها غير تالفة عند الولادة ويمكن أن تصنع الألدوستيرون لفترة من الوقت، لذلك تتطور أزمة إهدار الملح في نهاية المطاف بشكل تدريجي ومتغير أكثر من حالة فرط تنسج الكظرية الخلقي بسبب نقص إنزيم 21 - هيدروكسيليز.
يأتي معظمهم إلى الرعاية الطبية بين أسبوعين و 3 أشهر من العمر، وبعد فترة من قلة زيادة الوزن والقيء، وُجد أنهم يعانون من الجفاف، ونقص حاد في صوديوم الدم، وفرط بوتاسيوم الدم ، وحماض استقلابي ("أزمة أديسون أو نوبة كظرية"). يكون الرينين مرتفعاً وليس الألدوستيرون. توفي العديد من الأطفال الذين ولدوا بهذه الحالة قبل أن يتم التعرف على طريقة التشخيص لبدء العلاج المناسب. في بعض الحالات، تكون الحالة أكثر اعتدالًا مع ظهور علامات وأعراض نقص القشرانيات المعدنية والجلوكوكورتيكويد بعد شهور أو حتى سنوات (ظهور متأخر).
نقص الكورتيكويد السكري
عدم كفاية إنتاج الكورتيزول له عواقب عديدة. يترافق ارتفاع هرمون ACTH مع فرط تصبغ ملحوظ حتى في فترة حديثي الولادة. مما لا شك فيه أن استجابة الكورتيزول غير الكافية للإجهاد تسرع من التدهور مع تطور الجفاف، ويمكن أن تسبب نقص السكر في الدم، وتساهم في ارتفاع معدل الوفيات في الأطفال.
خلال تطور الجنين
ضعف إنتاج هرمون ديهيدرو ايبي آندروستيرون قبل الولادة من الغدد الكظرية الجنينية يؤدي إلى انخفاض مستويات هرمون الإستريول لدى الأمهات بشكل غير طبيعي بحلول منتصف الحمل. لا تزال آثار ضعف إنتاج البروجسترون من خلايا المشيمة التي تنشأ من الطفل المصاب (الأرومة المغذية) في حالة تضخم الغدة الكظرية الخلقي الدهني بسبب نقص إنزيم انشقاق سلسلة الكولسترول الجانبية (P450scc) غير واضحة، ولكن يُعتقد أنها تؤدي إلى إجهاض عندما يكون النقص في نشاط الإنزيم شديدًا بدرجة كافية. نتائج انخفاض أو غياب إنتاج هرمون التستوستيرون بواسطة خلايا لايديغ الجنينية في الذكر مفصلة أدناه.
المرضى الإناث
الإناث (الرموز الوراثية XX) المصابات بتضخم الغدة الكظرية الخلقي الشحمي يُولدنَ بتشريح الحوض الخارجي والداخلي الطبيعي. يأتون إلى الرعاية الطبية عندما يصابون بأزمة الغدة الكظرية التي تسبب إهدار الملح أو غيرها من علامات قصور الغدة الكظرية التقدمي.
مع استبدال الجلوكوكورتيكويد والقشرانيات المعدنية، تصل هؤلاء الفتيات إلى سن البلوغ. نظرًا لأن المبايض تكون غير نشطة نسبيًا في حياة الجنين والطفولة، فإنها تتعرض لضرر ضئيل من تراكم الدهون أثناء الطفولة. في حالة تضخم الغدة الكظرية الخلقي الشحمي بسبب نقص StAR، عندما ترتفع مستويات الجونادوتروبين تبدأ البلوغ، على الرغم من عدم كفاءة تخليق الستيرويد الجنسي، عادة ما ينتج المبيض استراديول كافٍ لإنتاج نمو الثدي، وفي بعض الحالات حتى الحيض، مع استمرار الحيض لبعض سنوات. يكون إنتاج الأندروجين في المبيض والغدة الكظرية ضئيلًا وينتج القليل من شعر العانة أو غيره من شعر الجسم.
ومع ذلك، يتم إنتاج استراديول وبروجسترون غير كافيين للحث على نضوج البويضة والإباضة. على الرغم من أن المبايض قبل البلوغ غير نشطة بدرجة كافية بحيث لا يتراكم أي دهون لإحداث ضرر، فبمجرد أن تبدأ في إنتاج هرمون الاستروجين، يبدأ تلف الدهون في التراكم وتتدهور القدرة على إنتاج الإستروجين، وكذلك الإباضة ببطء. تتشكل الأكياس أيضًا في المبايض. النساء المصابات بـ تضخم الغدة الكظرية الخلقي الشحمي يعانين من العقم على الأرجح بسبب الإباضة.
المرضى الذكور
الأعضاء التناسلية للأجنة XY مع تضخم الغدة الكظرية الخلقي الشحمي هي أقل من اللازم بسبب ضعف تخليق هرمون الستيرويد. تصنع خصيتان الجنين هرمون مضاد مولر AMH، الذي يمنع تكوين الرحم والمهبل الداخلي، ولكن نظرًا لأن خلايا لايديغ تفشل في إنتاج هرمون التستوستيرون أثناء التطور حتى في الاستجابةً لموجهة الغدد التناسلية المشيمائية hCG، تظل الخصيتان عادةً في البطن أو في القنوات الأربية (الخصيتين المعلقتين) وتكون غير وظيفية. وبالتالي، فإن مرضى XY لا يمرون بمرحلة البلوغ ويظلون يعانون من العقم.
بالإضافة إلى الخصيتين المتبقيتين بالداخل، فإن تكوين القضيب، الذي يعتمد أيضًا على هرمون التستوستيرون، معرض للخطر. ومن ثَمَّ، فإن الأعضاء التناسلية الخارجية في معظم الأطفال تشبه الأعضاء التناسلية للإناث الطبيعيات (على الرغم من أن المهبل عبارة عن كيس أعمى قصير)، أو غامض قليلاً (أنثى أكثر من الذكر). تم افتراض أن جميع حالات XY المبلغ عنها تقريبًا من الفتيات وتم تربيتها على هذا النحو.
ظهور الأشكال المتأخرة للمرض
تم وصف الحالات الأكثر اعتدالًا من تضخم الغدة الكظرية الخلقي الشحمي والتي تنشأ من طفرات أقل خطورة تؤثر على قدرة StAR على إنتاج الستيرويد ولكنها لا تلغيها.[1] في هذه الحالات، يظهر نقص القشرانيات المعدنية لعدة سنوات بعد الولادة. قد يكون إنتاج الستيرويد الجنسي كافياً للسماح بالتطور الجنسي الطبيعي وكذلك حتى الخصوبة.
يتم تشخيص هذه الأشكال غير الكلاسيكية للاضطراب أحيانًا على أنها من النوع 3 من عوز الجلوكوكورتيكويد العائلي.[2]
الوراثة
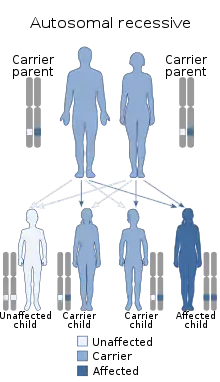
هذا المرض الوراثي متنحِ وراثياً (autosomal recessive). طُوِّر فهم الأساس الجزيئي له في العقد الماضي من خلال فهم أفضل لتكوين الستيرويد الكظري بالإضافة إلى الدراسات الجينية للمرضى المصابين.[3] كان من المفترض أن يكون تضخم الغدة الكظرية الخلقي الشحمي ناتجًا عن خلل في الإنزيم الذي حول الكوليسترول إلى البريغنينولون. يتم التوسط في تفاعلات التحويل بواسطة إنزيم واحد، كان يُشار إليه سابقًا باسم 20,22-desmolase، ولكن يُعرف الآن باسم سيتوكروم P450scc (إنزيم انشقاق سلسلة جانبية للكوليسترول). ومع ذلك، تم تحديد حالات قليلة من تضخم الغدة الكظرية الخلقي الشحمي بسبب طفرة وعيوب في P450scc. على الرغم من اعتبار الاضطراب صبغيًا جسديًا متنحيًا، إلا أن طفرة واحدة في P450scc يمكن أن تكون كافية للتسبب في الحالة.[4] وعُثِرَ على جميع حالات تضخم الغدة الكظرية الأخرى التي تمت دراستها بسبب طفرات في جين البروتين الأساسي الذي ينقل الكوليسترول إلى الميتوكوندريا، StAR، المشفر بواسطة جين على الكروموسوم 8p11.2 في الإنسان.
فرط تنسج الغدة الكظرية الخلقي هو عائلة من الأمراض الصبغية المتنحية الناتجة عن عيوب في خطوات تخليق هرمونات الستيرويد من الكوليسترول. تتضمن جميع أشكال تضخم الغدة الكظرية الخلقي إنتاجًا مفرطًا أو معيبًا للستيرويدات الجنسية ويمكن أن تمنع أو تضعف تطور الخصائص الجنسية الأولية أو الثانوية عند الرضع والأطفال والبالغين المصابين. يتضمن الكثير منها أيضًا الإنتاج المفرط أو المعيب للقشرانيات المعدنية، والتي يمكن أن تسبب ارتفاع ضغط الدم أو إهدار الملح.
يعد تضخم الغدة الكظرية الخلقي الشحمي أحد أكثر أشكال تضخم الغدة الكظرية الخلقي نادرًا وينتج عن عيوب في الخطوات من الكوليسترول إلى البريغنينولون.[3] يؤدي هذا إلى خسارة كارثية لمعظم أو كل هرمونات الستيرويد في الجسم. وهو ناتج عن طفرات في أي من البروتينين: السيتوكروم P450scc والبروتين التنظيمي الحاد الستيرويدي المنشأ (StAR).
الفيزيولوجيا المرضية
ينتج النقص عن اختلال في تخليق جميع الفئات الثلاث من المنشطات الكظرية (الكورتيزول والقشرانيات المعدنية والستيرويدات الجنسية) ومستويات عالية من الهرمون الموجه لقشر الكظر (ACTH). يستمر مستوى منخفض من تخليق الستيرويد حتى بدون النقل الفعال، ولكنه نادرًا ما يكون كافياً لمنع عواقب النقص. في حين أن الخسارة الشديدة في إنتاج الستيرويد تؤدي إلى ظهور المرض في غضون أسابيع قليلة من الولادة، فإن الأشكال الأكثر اعتدالًا (بداية متأخرة) يمكن أن تظهر بعد سنوات من الولادة. على عكس نماذج المرض في الفئران، لا يعاني المرضى المصابون بتضخم الغدة الكظرية الخلقي الشحمي دائمًا من تضخم الغدة الكظرية بسبب تراكم الدهون. قد يكون هذا جزئيًا بسبب استبدال الهرمون المستخدم لإبقائهم على قيد الحياة لمنع فرط تحفيز الغدة النخامية.
يحفز الهرمون الموجه لقشر الكظر نمو خلايا الغدة الكظرية ويزيد من مستقبلات LDL لتضخيم نقل الكوليسترول إلى خلايا قشرة الغدة الكظرية التي تصنع الستيرويدات الكظرية، حيث تتراكم حيث يمكن للقليل أن يدخل الميتوكوندريا للتحول إلى الستيرويد. عادة، تشير الستيرويدات الكظرية إلى وجودها إلى الدماغ لتعديل مستويات ACTH (تثبيط التغذية المرتدة). ومع ذلك، في حالة عدم وجود هذا، ترتفع مستويات ACTH ويستمر امتصاص الكوليسترول من قبل الخلايا القشرية دون إنقطاع. تتضخم الغدة الكظرية بشكل ملحوظ (فرط التنسج) بسبب تراكم الدهون. يُعتقد أن تراكم الدهون يؤدي إلى مزيد من الضرر للخلايا ("فرضية الإصابة الثانية").
لأن P450scc و StAR ضروريان أيضًا لتخليق الستيرويد الجنسي في الخصية والمبيض، فإن إنتاج هرمون التستوستيرون بواسطة خلايا لايديغ في الخصية والأندروجينات (مما يؤدي إلى إنتاج هرمون الاستروجين بواسطة الخلايا الحبيبية) والبروجستيرون بواسطة الخلايا القرابية المبيضية والخلايا الأصفرية، على التوالي، يمكن أن يضعف. على غرار الغدة الكظرية، يؤدي تراكم الكوليسترول إلى إتلاف خلايا لايديغ في الخصيتين. في المبيض، يبدأ الضرر بعد البلوغ، وهو الوقت الذي يبدأ فيه المبيض في صنع الستيرويد مع تطور الجريب. تصنع المشيمة أيضًا الستيرويد للمساعدة في الحفاظ على الحمل. ومع ذلك، نظرًا لأن StAR ليس مطلوبًا لإنتاج الستيرويد المشيمي، فإن الحمل ينتهي. عندما تكون الطفرة في P450scc التي تسبب تضخم الغدة الكظرية الخلقي الدهني إما متغايرة الزيجوت أو أن وجودها على كلا الأليلين لا يدمر جميع الوظائف تمامًا، يمكن للأطفال المصابين البقاء على قيد الحياة حتى الولادة أيضًا. وتجدر الإشارة أيضًا إلى أن تضخم الغدة الكظرية لا يوجد دائمًا في المريض، خاصة في الحالات التي يكون فيها سبب حدوث طفرة في جين P450scc.[5]
تختلف الفيزيولوجيا المرضية للتضخم الغدة الكظرية الخلقي الدهني الشحمي عن الأشكال الأخرى لـتضخم الغدة الكظرية الخلقي الدهني في جوانب معينة. أولاً، الجين المصاب في معظم الحالات هو بروتين النقل (StAR) بدلاً من إنزيم ستيرويدوجينيك. ثانيًا، لأن الخلل يضعف كل تخليق الستيرويد. وبالتالي، لا توجد مشاكل بسبب فرط القشرانيات المعدنية أو الأندروجينات. ثالثًا، يتسبب تراكم الدهون في إتلاف الخصيتين والمبيضين، بحيث لا يمكن الحفاظ على وظيفة الغدد التناسلية والخصوبة حتى مع الاستبدال المناسب لهرمون الغدة الكظرية (وفي حالة عدم وجود تدخل آخر).
تشخيص
من حيث تشخيص هذه الحالة، يمكن إجراء التسلسل الجيني.[6]
تدبير علاجي
إدارة أزمات إهدار الملح ومعالجة القشرانيات المعدنية هي مثل الأشكال الأخرى من تضخم الغدة الكظرية الخلقي الذي يهدر الملح: المحلول الملحي والفلودروكورتيزون. يمكن توفير القشرانيات السكرية بأدنى حد من الجرعات البديلة لأنه لا توجد حاجة لقمع فرط الأندروجينات الكظرية أو القشرانيات المعدنية. كما هو الحال مع الأشكال الأخرى من قصور الغدة الكظرية، هناك حاجة إلى زيادة الجلوكوكورتيكويد لتغطية الإجهاد.
المرضى الإناث
قد تحتاج XX الإناث المصابات بضخم الغدة الكظرية الخلقي الشحمي إلى استبدال الإستروجين عند البلوغ أو بعده. تم استخدام التدخل الفعال للحفاظ على إمكانية الخصوبة والحمل في الإناث المصابات بتضخم الغدة الكظرية الخلقي الشحمي.[7] في تقرير حالة في عام 2009، خضعت امرأة مصابة بـتضخم الغدة الكظرية الخلقي الشحمي المتأخر بسبب نقص StAR للعلاج بالهرمونات البديلة بالاشتراك مع تقنية الخصوبة المساعدة، حقن الحيوانات المنوية بالبويضة.[8] أدى ذلك إلى الإباضة وزرع البويضة الملقحة في المختبر، وهي ولادة ناجحة.
المرضى الذكور
يعاني معظم أطفال XY من سوء المعاملة لدرجة أنهم تربوا كبنات. الخصيتان غير وظيفيتين بشكل موحد وغير نزلتين؛ يتم إزالتها عندما يتم التشخيص بسبب خطر الإصابة بالسرطان في هذه الأنسجة.[9]
وبائيات
تضخم الغدة الكظرية الخلقي الشحمي نادر جدًا في سكان أوروبا وأمريكا الشمالية. تحدث معظم الحالات في اليابان وكوريا (حيث تبلغ نسبة الإصابة 1 من كل 300000 ولادة) وفلسطين. على الرغم من الوراثة الجسدية، كان هناك غلبة غير مفسرة للإناث الجينية في الحالات المبلغ عنها.[10]
انظر أيضًا
المراجع
- "Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia"، J. Clin. Endocrinol. Metab.، 91 (12): 4781–5، ديسمبر 2006، doi:10.1210/jc.2006-1565، PMC 1865081، PMID 16968793، مؤرشف من الأصل في 18 يناير 2021.
- "Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency"، J. Clin. Endocrinol. Metab.، 94 (10): 3865–3871، أكتوبر 2009، doi:10.1210/jc.2009-0467، PMC 2860769، PMID 19773404.
- "Phenotypic variations in lipoid congenital adrenal hyperplasia"، Pediatr Endocrinol Rev، 3 (3): 258–71، مارس 2006، PMID 16639391.
- "Heterozygous mutation in the cholesterol side chain cleavage enzyme (p450scc) gene in a patient with 46,XY sex reversal and adrenal insufficiency"، J Clin Endocrinol Metab، 86 (8): 3820–5، أغسطس 2001، doi:10.1210/jc.86.8.3820، PMID 11502818.
- "Severe combined adrenal and gonadal deficiency caused by novel mutations in the cholesterol side chain cleavage enzyme, P450scc"، J. Clin. Endocrinol. Metab.، 93 (3): 696–702، مارس 2008، doi:10.1210/jc.2007-2330، PMC 2266942، PMID 18182448.
- Kim, Chan Jong (ديسمبر 2014)، "Congenital lipoid adrenal hyperplasia"، Annals of Pediatric Endocrinology & Metabolism، 19 (4): 179–183، doi:10.6065/apem.2014.19.4.179، ISSN 2287-1012، PMC 4316413، PMID 25654062.
- "Phenotypic features of 46, XX females with StAR protein mutations"، Pediatr Endocrinol Rev، 5 (2): 633–41، ديسمبر 2007، PMID 18084157.
- "Conception and pregnancy outcome in a patient with 11-bp deletion of the steroidogenic acute regulatory protein gene"، Fertil Steril، 91 (3): 934.e15–8، مارس 2009، doi:10.1016/j.fertnstert.2008.07.1770، PMID 18829024.
- "Role of a founder c.201_202delCT mutation and new phenotypic features of congenital lipoid adrenal hyperplasia in Palestinians"، J Clin Endocrinol Metab، 92 (10): 4000–8، أكتوبر 2007، doi:10.1210/jc.2007-1306، PMID 17666473.
- Cantú-Reyna, Consuelo؛ Zepeda, Luis Manuel؛ Montemayor, René؛ Benavides, Santiago؛ González, Héctor Javier؛ Vázquez-Cantú, Mercedes؛ Cruz-Camino, Héctor (27 سبتمبر 2016)، "Incidence of Inborn Errors of Metabolism by Expanded Newborn Screening in a Mexican Hospital"، Journal of Inborn Errors of Metabolism and Screening، 4: 232640981666902، doi:10.1177/2326409816669027.
 بوابة طب
بوابة طب