Milroy's disease
Milroy's disease (MD) is a familial disease characterized by lymphedema, commonly in the legs, caused by congenital abnormalities in the lymphatic system. Disruption of the normal drainage of lymph leads to fluid accumulation and hypertrophy of soft tissues.[2][3]
| Milroy's disease | |
|---|---|
| Other names | Milroy disease, Nonne-Milroy-Meige syndrome, Hereditary lymphedema[1] |
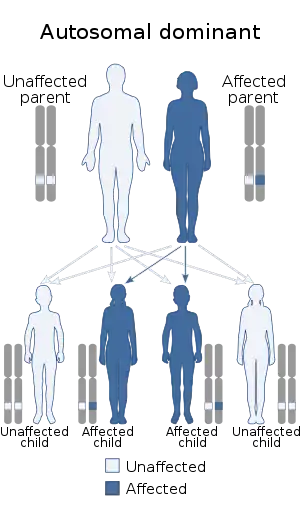 | |
| This condition is inherited in an autosomal dominant manner | |
| Specialty | Medical genetics |
It was named by Sir William Osler for William Milroy, a Canadian physician, who described a case in 1892, though it was first described by Rudolf Virchow in 1863.[4][5]
Presentation
The most common presentation of Milroy Disease is unilateral lower extremity lymphedema, and may also be accompanied by hydrocele. Males and females may have upslanting toenails, deep creases in the toes, wart-like growths (papillomas), and prominent leg veins. Some individuals develop non-contagious skin infections called cellulitis that can damage the thin tubes that carry lymph fluid (lymphatic vessels). Episodes of cellulitis can cause further swelling in the lower limbs.[6]
Genetics
This disease is more common in women and an association with the gene FLT4 has been described.[7] FLT4 codes for VEGFR-3, which is implicated in development of the lymphatic system.

Milroy's disease is also known as primary or hereditary lymphedema type 1A or early onset lymphedema. It is a very rare disease with only about 200 cases reported in the medical literature. Milroy's disease is an autosomal dominant condition caused by a mutation in the FLT4 gene which encodes of the vascular endothelial growth factor receptor 3 (VEGFR-3) gene located on the long arm (q) on chromosome 5 (5q35.3).[8]
In contrast to Milroy's disease (early onset lymphedema type 1A,) which typically has its onset of swelling and edema at birth or during early infancy, hereditary lymphedema type II, known as Meige disease, has its onset around the time of puberty. Meige disease is also an autosomal dominant disease. It has been linked to a mutations in the ‘forkhead’ family transcription factor (FOXC2) gene located on the long arm of chromosome 16 (16q24.3). About 2000 cases have been identified. A third type of hereditary lymphedema, that has an onset after the age of 35 is known as lymph-edema tarda.[8]
Diagnosis
Only conservative measures can be taken. Certain treatments for lymphedema disorders may possibly alleviate specific symptoms; no cure and it is usually congenital. Genetic counseling can be done. May have similar health conditions, delays, disorders, and physical traits associated with other lymphatic genetic diseases and chromosome #5 abnormalities.
Prognosis
MD does not normally affect life expectancy.[9]
Medscape states patients may have recurrent streptococcal cellulitis and lymphangitis, with subsequent hospitalizations for antibiotic therapy. A rare complication is the appearance of lymphangiosarcoma or angiosarcoma in patients with persistent lymphedema. Some patients may develop protein-losing enteropathy and visceral involvement. Chylous ascites and chylothorax rarely occur.
See also
References
- Bolognia JL, Jorizzo JL, Rapini RP (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1. OCLC 1058487222.
- James WD, Berger TG, Elston DM, Andrews GC, Odom RB (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. p. 849. ISBN 978-0-7216-2921-6. OCLC 937244604.
- Strayer DL, Rubin R (2007). Rubin's Pathology: Clinicopathologic Foundations of Medicine (5th ed.). Hagerstwon, MD: Lippincott Williams & Wilkins. ISBN 978-0-7817-9516-6.
- synd/1326 at Who Named It?
- Milroy WF (1892). "An undescribed variety of hereditary edema". New York Medical Journal. 56: 505–8.
- "Milroy Disease". United States Library of Medicine. Retrieved 1 March 2014.
- Spiegel R, Ghalamkarpour A, Daniel-Spiegel E, Vikkula M, Shalev SA (2006). "Wide clinical spectrum in a family with hereditary lymphedema type I due to a novel missense mutation in VEGFR3". Journal of Human Genetics. 51 (10): 846–50. doi:10.1007/s10038-006-0031-3. PMID 16924388.
- "Hereditary Lymphedema". Retrieved 1 September 2016.
- Rockson SG (October 2010). "Causes and consequences of lymphatic disease". Annals of the New York Academy of Sciences. 1207 Suppl 1: E2-6. Bibcode:2010NYASA1207E...2R. doi:10.1111/j.1749-6632.2010.05804.x. PMID 20961302.
Further reading
- Brice, Glen W.; Mansour, Sahar; Ostergaard, Pia; Connell, Fiona; Jeffery, Steve; Mortimer, Peter (1993). "Milroy Disease". GeneReviews®. University of Washington, Seattle. Retrieved 15 March 2019.