Rubinstein–Taybi syndrome
Rubinstein–Taybi syndrome (RTS) is a rare genetic condition characterized by short stature, moderate to severe learning difficulties, distinctive facial features, and broad thumbs and first toes.[2] Other features of the disorder vary among affected individuals. These characteristics are caused by a mutation or deletion in the CREBBP and/or EP300 gene located on chromosome 16.
| Rubinstein–Taybi syndrome | |
|---|---|
| Other names | Broad thumb-hallux syndrome or Rubinstein syndrome[1] |
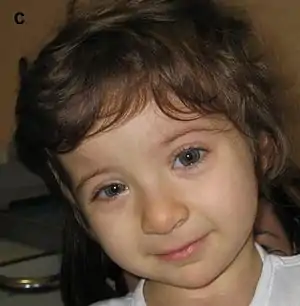 | |
| Child with Rubinstein–Taybi syndrome | |
| Specialty | Medical genetics |
People with this condition have an increased risk of developing noncancerous and cancerous tumors, leukemia, and lymphoma. This condition is sometimes inherited as an autosomal dominant pattern and is uncommon. Many times it occurs as a de novo (not inherited) occurrence. It occurs in an estimated 1 in 125,000-300,000 births.
Presentation
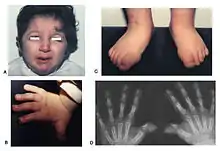
Rubinstein–Taybi syndrome presents itself from birth, and is usually hallmarked by delayed physical and cognitive growth.
Typical features of the disorder include:
- Broad thumbs and broad first toes and clinodactyly of the 5th finger[3]
- Mental disability
- Small height, low bone growth, small head
- Cryptorchidism in males
- Unusual facies involving the eyes, nose, and palate
- Anesthesia may be dangerous in these patients: "According to the medical literature, in some cases, individuals with Rubinstein–Taybi syndrome may have complications (e.g., respiratory distress and/or irregular heart beats [cardiac arrythmias]) associated with a certain muscle relaxant (succinylcholine) and certain anesthesia. Any situations requiring the administration of anesthesia or succinylcholine (e.g., surgical procedures) should be closely monitored by skilled professionals (Anesthesiologists)."[4] Primary literature suggests the children may have a higher rate of cardiac physical and conduction abnormalities which may cause unexpected results with cardioactive medications.[5] A further editorial reply in the British Journal of Anaesthesia discusses changes in the face and airway structure making it more difficult to secure the airway under anaesthesia, however, complications appeared in a minority of cases, and routine methods of airway control in the operating room appears to be successful. They recommended close individual evaluation of Rubinstein–Taybi patients for anaesthetic plans.[6]
A 2009 study found that children with RTS were more likely to be overweight and to have a short attention span, motor stereotypies, and poor coordination. The study hypothesized that the identified CREBBP gene impaired motor skills learning.[7] Other research has shown a link with long-term memory (LTM) deficit.[8][9]
It is diagnosed when a heterozygous pathogenic variant of the CREBBP gene is identified in the individual. It exhibits an autosomal dominant inheritance pattern, but some documented cases show heterozygous individuals exhibiting germline mosaicism. This condition affects men and women equally, and is often misdiagnosed with other diseases or disabilities that result in delayed mental development.
Genetics
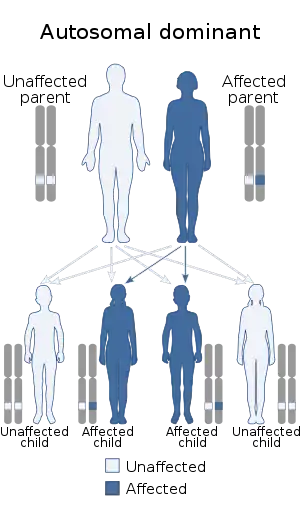
Rubinstein–Taybi syndrome is a microdeletion syndrome involving chromosomal segment 16p13.3 and is characterized by mutations in the CREBBP gene.[10][11] Varying amounts of material are deleted from this section of the chromosome and accounts for the spectrum of physiological symptoms.[12]
The CREBBP gene makes a protein that helps control the activity of many other genes. The protein, called CREB-binding protein, plays an important role in regulating cell growth and division and is essential for normal fetal development. If one copy of the CREBBP gene is deleted or mutated, cells make only half of the normal amount of CREB binding protein. A reduction in the amount of this protein disrupts normal development before and after birth, leading to the signs and symptoms of Rubinstein–Taybi syndrome.[13]
Mutations in the EP300 gene are responsible for a small percentage of cases of Rubinstein–Taybi syndrome. These mutations result in the loss of one copy of the gene in each cell, which reduces the amount of p300 protein by half. Some mutations lead to the production of a very short, nonfunctional version of the p300 protein, while others prevent one copy of the gene from making any protein at all. Although researchers do not know how a reduction in the amount of p300 protein leads to the specific features of Rubinstein–Taybi syndrome, it is clear that the loss of one copy of the EP300 gene disrupts normal development.
A mouse model has been identified in order to perform experimental research procedures. The model has exhibited the same clinical symptoms seen in humans and has become a foundation for future research.[14]
Unfortunately, in nearly 40% of cases, neither gene, CREBBP or EP300, can be implicated in the cause of the disease. In these cases, there is no mutation on the 16th chromosome leaving many more questions yet to be answered. However, new research shows that around 55% of cases involve a mutation in the CBP gene, further suggesting that this disease has its origins in mutation of the regulatory mechanisms of transcription (Izumi, 2016).[15]
Treatment
There is no existing treatment that reverses or cures RTS. There are, however, ways to manage and reduce symptoms for patients. Patients with RTS suffer from a diverse breadth of symptoms. These include cognitive-developmental impairment, heart abnormalities, delayed bone growth and skeletal abnormalities, auditory impairment, urinary tract abnormalities, including kidney problems, and dental and speech problems. Not every patient will suffer from all or multiple symptoms, and not every patient will experience the same symptoms, meaning they differ from patient to patient. Due to there being a wide range of symptoms, RTS patients are referred to specialists that focus on each specific symptom. There is not a specialist for RTS. For example, patients will go to an orthopedic surgeon and physical therapy for skeletal and growth abnormalities, like scoliosis but will go to a cardiologist if they suffer from heart abnormalities or a dentist if they suffer from dental abnormalities. Individuals suffering from cognitive developments usually are part of special education programs and speech therapy. The specialists the individuals go to match the symptoms the individuals have. Regular check-ups and monitoring are needed for cardiac, dental, auditory, and renal abnormalities. Genetic counseling is also recommended for affected individuals and their families.[16]
History
Rubinstein–Taybi syndrome was first unofficially mentioned in a French orthopedic medical journal in 1957 by Greek physicians' doctors: Michail, Matsoukas, and Theodorou. The medical journal reported a case concerning a seven-year-old boy with radically deviated/arched thumbs, long nose, muscular hypotonia, along with physical and mental underdevelopment. At this point in time the case study mentioned by the Greek physicians was considered to be an anomaly due to the fact that there hadn't been any other reported cases of children with these specific physical and mental characteristics. The doctors accredited with discovering the syndrome and therefore bear its name-sake were unaware of this journal at the time of their discovery. However, it is acknowledged that the 1957 case reported in the French journal of orthopedic medicine is most likely the first reported case of RTS.
Dr. Jack Herbert Rubinstein, an American pediatrician reported assessing a three-year-old girl with unusual facial and digital findings in 1958. Similarly, that same year Rubinstein had evaluated another child with similar characteristics, this time a seven-year-old boy. Having sensed a striking similarity between these two unrelated cases Rubinstein tried distributing photos and information concerning these two cases to other clinics in the U.S. from 1959 to 1960. Rubinstein graduated from Harvard Medical School and worked as the director of the Hamilton County Diagnostic Clinic for the Mentally Retarded. He has worked in behavioral and developmental pediatrics for many years prior to the discovery of this new syndrome.
In 1961, Dr. Hooshang Taybi, an Iranian-American pediatric radiologist, reported having assessed a three-year-old boy that appeared to have the same syndrome as described by Rubinstein. During the summer of 1963 Dr. Taybi reported having evaluated seven children with characteristics such as broad thumbs and great toes, "unusual" facial features, and intellectual disabilities – these findings went on to appear in the American Journal of Diseases of Children documenting these characteristics as a syndrome. Dr. Hooshang Taybi graduated from Tehran University School of Medicine and worked for the Ministry of Health. Later in his career he taught and practiced pediatric radiology in Oklahoma and Indiana. He had identified three new syndromes with his colleagues, among them is Rubinstein–Taybi syndrome.
In 1992 the first genetic abnormalities that act as markers for Rubinstein-Taybi syndrome were identified. These abnormalities are said to affect either chromosome 16 or chromosome 22. The specific chromosome impacted by a mutation determines the type of Rubinstein–Taybi syndrome that may occur. A mutation of the CREBP gene on chromosome 16 gives rise to the first form of RTS (most common). While a mutation of the EP300 gene on chromosome 22 is characteristic of the second form of RTS.
References
- Online Mendelian Inheritance in Man (OMIM): Rubinstein–Taybi syndrome - 180849
- Petrij, F; Dauwerse, HG; Blough, RI; Giles, RH; van der Smagt, JJ; Wallerstein, R; Maaswinkel-Mooy, PD; van Karnebeek, CD; van Ommen, GJ; van Haeringen, A; Rubinstein, JH; Saal, HM; Hennekam, RC; Peters, DJ; Breuning, MH (March 2000). "Diagnostic analysis of the Rubinstein-Taybi syndrome: five cosmids should be used for microdeletion detection and low number of protein truncating mutations". Journal of Medical Genetics. 37 (3): 168–76. doi:10.1136/jmg.37.3.168. PMC 1734540. PMID 10699051.
- Hennekam RC (Sep 2006). "Rubinstein-Taybi syndrome". Eur J Hum Genet. 14 (9): 981–985. doi:10.1038/sj.ejhg.5201594. PMID 16868563.
- "Anesthesia". Archived from the original on 2011-10-18. Retrieved 2012-04-11.
- Stirt JA (July 1981). "Anesthetic problems in Rubinstein-Taybi syndrome". Anesthesia and Analgesia. 60 (7): 534–6. doi:10.1213/00000539-198107000-00016. PMID 7195672. S2CID 37522638.
- Dearlove OR, Perkins R (March 2003). "Anaesthesia in an adult with Rubinstein-Taybi syndrome". British Journal of Anaesthesia. 90 (3): 399–400, author reply 399–400. doi:10.1093/bja/aeg537. PMID 12594162.
- Galéra C, Taupiac E, Fraisse S, et al. (2009). "Socio-behavioral sharacteristics of children with Rubinstein–Taybi syndrome". J Autism Dev Disord. 39 (9): 1252–1260. doi:10.1007/s10803-009-0733-4. PMID 19350377. S2CID 5456561.
- Bourtchouladze R, Lidge R, Catapano R, et al. (September 2003). "A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4". Proceedings of the National Academy of Sciences of the United States of America. 100 (18): 10518–22. Bibcode:2003PNAS..10010518B. doi:10.1073/pnas.1834280100. JSTOR 3147748. PMC 193593. PMID 12930888.
- Alarcón JM, Malleret G, Touzani K, et al. (June 2004). "Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration". Neuron. 42 (6): 947–59. doi:10.1016/j.neuron.2004.05.021. PMID 15207239. S2CID 15669747.
- Wójcik, C; Volz, K; Ranola, M; Kitch, K; Karim, T; O'Neil, J; Smith, J; Torres-Martinez, W (February 2010). "Rubinstein-Taybi syndrome associated with Chiari type I malformation caused by a large 16p13.3 microdeletion: a contiguous gene syndrome?". American Journal of Medical Genetics Part A. 152A (2): 479–83. doi:10.1002/ajmg.a.33303. PMID 20101707. S2CID 205312346.
- Petrij F, Giles RH, Dauwerse HG, et al. (July 1995). "Rubinstein–Taybi syndrome caused by mutations in the transcriptional co-activator CBP". Nature. 376 (6538): 348–51. Bibcode:1995Natur.376..348P. doi:10.1038/376348a0. PMID 7630403. S2CID 4254507.
- Reference, Genetics Home. "Rubinstein-Taybi syndrome". Genetics Home Reference. Retrieved 2020-05-06.
- Milani, Donatella; Manzoni, Francesca Maria Paola; Pezzani, Lidia; Ajmone, Paola; Gervasini, Cristina; Menni, Francesca; Esposito, Susanna (2015-01-20). "Rubinstein-Taybi syndrome: clinical features, genetic basis, diagnosis, and management". Italian Journal of Pediatrics. 41 (1): 4. doi:10.1186/s13052-015-0110-1. ISSN 1824-7288. PMC 4308897. PMID 25599811.
- Oike, Y.; Hata, A.; Mamiya, T.; Kaname, T.; Noda, Y.; Suzuki, M.; Yasue, H.; Nabeshima, T.; Araki, K.; Yamamura, K. (March 1999). "Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism". Human Molecular Genetics. 8 (3): 387–396. doi:10.1093/hmg/8.3.387. ISSN 0964-6906. PMID 9949198.
- Izumi, K (2016). "Disorders of Transcriptional Regulation: An Emerging Category of Multiple Malformation Syndromes". Molecular Syndromology. 7 (5): 262–273. doi:10.1159/000448747. PMC 5109993. PMID 27867341. Retrieved 3 November 2021.
- "Rubinstein Taybi Syndrome". NORD (National Organization for Rare Disorders). Retrieved 2020-05-06.