Dinámica química
La dinámica química es una parte de la química física que estudia las interacciones entre las partículas que conforman las diferentes sustancias que intervienen en una reacción química y también los estudios sobre transporte y difusión de las sustancias químicas en un sistema o en la naturaleza.
La dinámica química es una de las tres áreas de la química según el Informe Westheimer (1965), junto a la química estructural y la química de síntesis. Su objetivo es el análisis de los procesos químicos desde una dimensión evolutiva, es decir, se encarga del estudio de la materia en el proceso de cambio químico y las causas de dichos procesos.
Las interacciones entre átomos, moléculas e iones son la causa explicativa de muchos fenómenos estudiados por esta rama de la química, como las propiedades de los sólidos, líquidos y gases; las transiciones de fase; la geometría durante las colisiones entre partículas; el estado de transición, los mecanismos de reacción, el equilibrio químico, etc.
Wilhelm Ostwald y Jacobus Henricus van 't Hoff son algunos de los científicos que desarrollaron inicialmente esta ciencia. Este último mereció el Premio Nobel en 1901 "por el descubrimiento de las leyes de la dinámica química y de la presión osmótica en las soluciones químicas"
En esta obra sobre dinámica química (posteriormente llamada cinética química) se ocupa de las velocidades de reacción y su relación con la energía libre.[1]
Diferentes enfoques de la dinámica química
Al tratar de responder a la pregunta ¿Cómo ocurren las reacciones?, la dinámica química adopta diferentes puntos de vista.[2]
Enfoque microscópico
Se encarga del estudio de los átomos y moléculas en el curso de una transformación química[3] y del análisis sistemático de las reacciones y la reactividad: velocidad, energía, mecanismos de reacción y equilibrio en las transformaciones químicas.[4]
Se divide a su vez en tres grandes áreas de estudio: termodinámica química, cinética química y mecánica estadística.[5]
Algunos de los temas de estudio desde este enfoque son:
- Termodinámica de los procesos irreversibles.
- Cinética química
- Teoría del transporte y difusión.
- Procesos no lineales.
- Sistemas ideales y sistemas de partículas en interacción.
- Química de superficies.
- Métodos de cálculo de la Dinámica molecular semiclásica y de Monte Carlo.
- Mecánica estadística de sistemas en equilibrio y no-equilibrio

Enfoque macroscópico
Se ocupa del estudio del transporte y evolución de sustancias químicas en el medio ambiente, generalmente a gran escala.[6]
Son ejemplos típicos de este enfoque los siguientes:
- Evolución de contaminantes. Procesos físico-químicos que sufren los plaguicidas en la naturaleza; su persistencia, actividad específica y destino final.[7]
- Distribución e impacto de las sustancias químicas vertidas en ecosistemas acuáticos o en la atmósfera.
Enfoque teórico cuántico
Estudia el comportamiento de las partículas (átomos, moléculas, ion) durante las reacciones químicas, especialmente el análisis del estado de transición y el camino de reacción, empleando para ello métodos teóricos y experimentales.[8]
1. Métodos teóricos en Dinámica química:
- Métodos cuánticos y cuasiclásico de trayectorias.[9]
- Teorías sobre el Estado de Transición:
- Teorías convencionales: teoría canónica, teoría termodinámica y teoría microcanónica.
- Teorías variacionales: camino de reacción de mínima energía
2. Técnicas experimentales:
- Espectroscopía y Dinámica de estados altamente excitados vibracionalmente.
Hacia 1950, John C. Polanyi estudió la transición de las moléculas durante una reacción desde un estado de alta excitación hasta un estado de menos excitación. Los cambios afectaban al grado de vibración y rotación de las moléculas. Si los productos de la reacción pasaban de estados de vibración extremadamente alta a estados de vibración extremadamente baja, se producían radiaciones de alta frecuencia. En 1958 Polanyi observó pequeños cambios vibracionales con radiación de baja frecuencia, introduciendo átomos de hidrógeno en una corriente de cloro gaseoso a baja presión. La reacción generó cloruro de hidrógeno en un estado de vibración excitado a muy alta temperatura y podía ser observada por un detector de emisión de infrarrojos. Esta luz se puede utilizar para producir un láser químico. Tras años de experimentos, Polanyi midió los estados de energía de transición, tanto los de vibración como los de rotación, de las moléculas del producto.
- Excitación de sobretonos en IR.
- Fotoextracción de electrones.
- Empleo de láser químico, desarrollados en 1965 por Kasper y George Claude Pimentel tras el desarrollo teórico de John C. Polanyi.
- Espectroscopía de moléculas transitorias en matrices inertes.
3. Métodos multivariacionales para cálculo teórico de estados excitados electrónicos:
- Método CASSCF
- Método CASPT2
- Técnicas de simulaciones por ordenador basadas en métodos estadísticos: método de dinámica molecular, método Monte Carlo.
Al estar involucrados trillones de partículas, los cálculos en el campo de la Dinámica molecular son muy complejos y requieren el uso de métodos aproximados, cálculos repetitivos y simulaciones por ordenador, y el empleo de orbitales simplificados como los orbitales moleculares de tipo Slater (OTS) los orbitales tipo Gaussianos (OTG).
Conceptos importantes en dinámica química
Potenciales de interacción
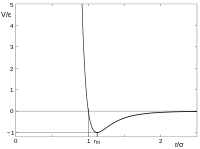
Proceden de la superposición entre las energías cinética y potencial de las partículas. La energía cinética tiende a dispersar las partículas formando un gas desordenado. La energía potencial eléctrica muestra las atracciones entre partículas y, por tanto, la tendencia al orden y la formación de estados condensados de la materia.
El potencial de Lennard-Jones es un ejemplo de estudio de esos potenciales.[10]
Los potenciales de interacción[11] entre átomos y moléculas parten de las siguientes hipótesis simplificadoras:
- Si interactúan dos átomos o moléculas entre sí, supondremos que no hay distorsión en las densidades electrónicas de esas partículas.
- Las fuerzas de Coulomb entre los electrones y núcleos se calculan usando las densidades electrónicas, dejando aparte otros fenómenos que contribuyen a la energía total como los efectos del intercambio electrónico, y la energía cinética, cuyos efectos se evalúan mediante una aproximación de un gas de electrones libres.
- Para el estudio de las densidades electrónicas de cada átomo se eligen las funciones de onda de Hartree-Fock.
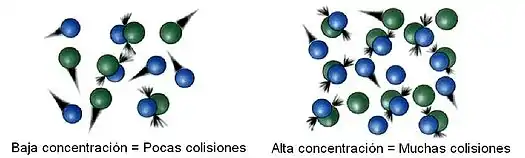
Dinámica de colisiones o choques
La teoría de colisiones explica cómo se acercan e interaccionan las partículas implicadas en una reacción química. Por simplicidad se escoge una reacción bimolecular con la formación del complejo activado.[12]
Funciones de partición
En mecánica estadística, la función de partición es una magnitud que reúne las propiedades estadísticas de un sistema en equilibrio termodinámico. Es una función de la temperatura y otros parámetros, como el volumen de un gas. La mayoría de las variables termodinámicas agregadas del sistema tales como la energía total, energía libre de Gibbs, entropía, y presión, pueden expresarse en términos de la función de partición o de sus derivadas.

Hay realmente varios tipos de funciones devpartición. La función de partición canónica se aplica a un conjunto canónico para el que suponemos que está en contactotérmicocon elexterior, tiene una temperatura T y posee a la vez un volumen y un número de partículas fijos. Si llamamos s (s = 1, 2, 3, ...) a los estados (microestados o estados cuánticos discretos) que elsistema puede ocupar y llamamos Es la energía total del sistema cuando está en ese microestado.
La función de partición es

donde definimos la "temperatura inversa" β como

siendo kB la constante de Boltzmann.
La función de partición está relacionada con las propiedades termodinámicas y tiene un significado estadístico importante.
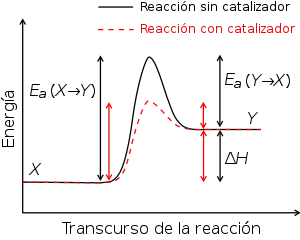
Teoría del estado de transición
Tras el desarrollo del concepto de superficies de energía potencial, varios teóricos como Evans, Polanyi y Eyring desarrollaron teorías cuantitativas de las velocidades de reacción basadas en la idea de que la formación del estado de transición controla la velocidad de la reacción. La especie química que existe en la región del estado de transición de la coordenada de reacción se llamó “complejo activado”, por lo que esta teoría se conoce también como la teoría del complejo activado.
Se basa en tres postulados:
- El sistema reaccionante al pasar del estado inicial al final, debe atravesar una región llamada estado de transición, cuya energía potencial corresponde a la energía más alta en la trayectoria de reacción.
- La especie química en el estado de transición se encuentra en equilibrio con los reactivos.
- La velocidad de reacción es igual al producto de las concentración de la especie del estado de transición y la frecuencia con que esta especie pasa a productos.
Reacciones unimoleculares
Los estudios experimentales de las reacciones unimoleculares han cambiado progresivamente. Los primeros trabajos usaban sistemas térmicos para excitar las moléculas. Después se introdujo la técnica de activación química que permite formar especies altamente excitadas que posteriormente puede isomerizar, descomponerse o bien perder energía mediante colisiones. Más tarde se desarrollaron métodos para preparar moléculas con energías concretas o en estados definidos, y fue posible la medida de las velocidades de reacción para tiempos cortos. Hoy en día, lae xcitación mediante láseres permite situar moléculas por encima de sus límites de disociación y seguir la reacción en tiempos de hasta 10-12segundos.
Los modernos métodos computacionales permiten conocer la geometría, energía y frecuencias de vibración de la molécula en función de la coordenada de reacción, lo que permite calcular las velocidades de reacción mediante modelos teóricos.[13]
Transiciones adiabáticas y no-adiabáticas
La dinámica adiabática considera que las interacciones entre átomos se pueden representar por una superficie de energía potencial. Esta es la aproximación de Born-Oppenheimer introducida por Born y Oppenheimer en 1927. Las primeras aplicaciones de este método fueron realizadas por Rice e Ramsperger en 1927 e Kassel
La teoría RRKM (1952) de Marcus fue desarrollada a partir de la teoría de transición de estado de Eyring en 1935. Estos métodos permiten calcular estimaciones de la velocidad de reacción a partir de características de la superficie de potencial.
La dinámica no adiabática considera las interacciones entre diversas superficies de energía potencial acopladas, correspondientes a diferentes estados cuánticos electrónicos de la molécula. Stueckelberg
(1932), Landau (1932) e Zener (1933) realizaron estudios que culminaron con la transición de Landau-Zener y permiten calcular la probabilidad de transición de las dos curvas cuando no se cumple la aproximación de Born-Oppenheimer.
Procesos dinámicos en superficies
Algunos fenómenos dinámicos muy relevantes ocurren en la superficie de los sólidos (catálisis, adsorción...), por lo que resulta de interés un análisis de los procesos de interacción superficie-medio externo y conocer los fundamentos físicos que explican los procesos de transporte cuántico de carga y de espín en nanoestructuras.
La física de superficies ha tenido un gran desarrollo gracias a las nuevas técnicas experimentales como las técnicas ópticas no lineales que han mostrado una gran sensibilidad a las condiciones estructurales de la superficie así como a los procesos que allí ocurren.
La microscopía de electrones (reflejados o fotoemitidos) es un ejemplo de técnica capaz de estudiar las superficies a escala nanométrica y determinar procesos dinámicos en superficies, como las transiciones de fase, los procesos de crecimiento o el estudio de la imanación.
Reacciones químicas en disolución. El efecto del disolvente
Las reacciones de isomerización y descomposición son unimoleculares y han sido estudiadas mediante técnicas de simulación por ordenador para comprender la influencia dinámica del disolvente sobre importantes fenómenos como la transferencia intermolecular de energía o la reactividad.
Algunas técnicas experimentales para el estudio dinámico de las reacciones en disolución son: técnicas de flujo, métodos de relajación, métodos con iniciación mediante radiaciones intensas y espectroscopias de resonancia.
Teoría de Kramers
Esta teoría fue enunciada por el físico holandés Hendrik Anthony Kramers en 1940 pero no se aceptó hasta 1980 por la ausencia de comprobación experimental con los medios de la época. Permite explicar algunos aspectos de las reacciones químicas en fase condensada como por ejemplo, las velocidades de las reacciones en disolución a partir de la difusión browniana en un campo de fuerzas, lo que supone que el movimiento aleatorio molecular es a la vez promotor e impedimento del movimiento.
Esta teoría describe el coeficiente de difusión mutua como una función del parámetro de movilidad de cadena, la masa molecular y la concentración. La teoría está basada en un proceso de difusión contra una barrera de potencial.
Distingue dos regímenes:
- Baja viscosidad: La constante de velocidad k crece con la viscosidad debido a los choques con el medio.
- Alta viscosidad: La constante k decrece con la velocidad.[14]
Modelos de reacciones de transferencia de electrones en disolución
Henry Taube clasificó los mecanismos de las reacciones redox en mecanismos de esfera interna (o de transferencia de átomos) y mecanismos de esfera externa (o de transferencia electrónica simple). La teoría para este último tipo de reacciones está bien establecida y se puede llegar a calcular las constantes de velocidad en algunos casos. Esta teoría está basada en el principio de Franck-Condon y en el hecho de que las transferencias de electrones entre átomos con niveles de energía acoplados son más fáciles (fenómeno cuántico de tunnelling, consecuencia del principio de incertidumbre de Heisenberg).[15]
La ecuación de Marcus proporciona la energía libre de Gibbs de activación para una reacción de transferencia de electrones en especies en disolución. Es la expresión cuantitativa de la teoría de Marcus, desarrollada por Rudolph Arthur Marcus, que permite a los científicos predecir el patrón y la velocidad de dichas reacciones.
- 1. Las moléculas D (donante) y A (aceptora) difunden a través de la disolución y forman un complejo precursor DA.

- 2. Transferencia de electrones en el complejo DA (de la molécula donante D a la molécula aceptora A) para formar el complejo D+A-

- 3. El complejo D+A- se rompe y las moléculas resultantes difunden

Véase también
Referencias
- Gran Enciclopedia Rialp, 1991.
- Chemical dynamics: a current review; National Research Council (U.S.). Panel on Chemical Dynamics. Washington, 1966.
- Química teórica y computacional. Juan Andrés y Juan Bertrán (editores). Univ. Jaime I. Castellón, 2000.
- Chemical dynamics. Joseph B. Dence, Harry B. Gray, George S.Hammond.California Institute of Technology. Nueva York, 1968.
- Dynamique chimique - Thermodynamique, cinétique et mécanique statistique. Grégoire Nicolis. Editeur Dunod, 2005.
- Dinámica química: transporte y comportamiento de sustancias químicas en el ambiente; un problema en salud ambiental. Freed, Virgil H. Centro Agronómico Tropical de Investigación y Enseñanza. Control integrado de plagas en sistemas de producción de cultivos para pequeños agricultores. Turrialba, CATIEUC/USAID/OIRSA, 1976. p.48-64.
- Chemical dynamics in fresh water ecosystems. Frank A. P. C. Gobas, John Alex McCorquodale. Lewis Publishers, 1992.
- An introduction to theoretical chemistry. Jack Simons. Cambridge University Press, 2003. También recibe por ello el nombre de Dinámica molecular.
- Principios básicos de química. Harry B. Gray, Gilbert P. Haight. Reverté, Barcelona, 1981 (reimpresión 2003)
- Dinámica Molecular 2D con un Potencial de Interacción tipo Lennard-Jones. Jerónimo Terrones Portas, 2007. Tomado de (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).
- Estudios en Dinámica química. Interacciones entre átomos de Capa Cerrada. Humberto Palza C.
- Teoría de las reacciones bimoleculares. Tomado de
- DINÁMICA DE REACCIONES UNIMOLECULARES EN FASE GAS. DESVIACIONES DEL COMPORTAMIENTO ESTADÍSTICO. Emilio Martínez-Núñez y Saulo A. Vázquez. Química Nova vol.25 no.4 Sao Paulo 2002
- Kramers. Physica, 7 (1940) 284-304.
- Química inorgánica, Volumen 2. Duward F. Shriver, P.W. Atkins y C.H. Lahgford. Editorial Reverté. Barcelona, 1998.
Enlaces externos
- Principles of Chemical Dynamics.
- Química teórica y computacional. Juan Andrés y Juan Bertrán (editores). Univ. Jaime I. Castellón, 2000.
| Control de autoridades |
|
|---|
 Datos: Q5807497
Datos: Q5807497