Anticuerpo
Los anticuerpos, (en la ciencia inmunoglobulinas Ig) son glucoproteínas del tipo gamma globulina. Pueden encontrarse de forma soluble en la sangre u otros fluidos corporales de los vertebrados, disponiendo de una forma idéntica que actúa como receptor de membrana en los linfocitos B y son empleados por el sistema inmunitario para identificar y neutralizar elementos extraños tales como bacterias y virus.[1]
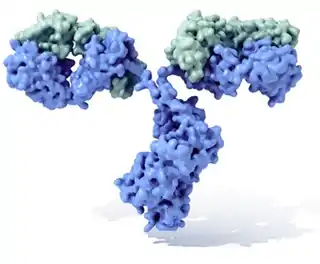
El anticuerpo típico está constituido por dos unidades estructurales básicas, cada una de ellas con dos grandes cadenas pesadas y dos cadenas ligeras de menor tamaño, que forman, por ejemplo, monómeros con una unidad, dímeros con dos unidades o pentámeros con cinco unidades. Los anticuerpos son sintetizados por un tipo de leucocito denominado linfocito B. Existen distintas modalidades de anticuerpo, isotipos, basadas en la forma de cadena pesada que posean. Se conocen cinco clases diferentes de isotipos en mamíferos que desempeñan funciones diferentes, contribuyendo a dirigir la respuesta inmune adecuada para cada distinto tipo de cuerpo extraño que encuentran.[2]
Aunque la estructura general de todos los anticuerpos es muy semejante, una pequeña región del ápice de la proteína es extremadamente variable, lo cual permite la existencia de millones de anticuerpos, cada uno con un extremo ligeramente distinto. A esta parte de la proteína se la conoce como región hipervariable. Cada una de estas variantes se puede unir a una "diana" distinta, que es lo que se conoce como antígeno.[3] Esta enorme diversidad de anticuerpos permite al sistema inmune reconocer una diversidad igualmente elevada de antígenos. La única parte del antígeno reconocida por el anticuerpo se denomina epítopo. Estos epítopos se unen con su anticuerpo en una interacción altamente específica que se denomina adaptación inducida, que permite a los anticuerpos identificar y unirse solamente a su antígeno único en medio de los millones de moléculas diferentes que componen un organismo.
El reconocimiento de un antígeno por un anticuerpo lo marca para ser atacado por otras partes del sistema inmunitario. Los anticuerpos también pueden neutralizar sus objetivos directamente, mediante, por ejemplo, la unión a una porción de un patógeno necesaria para que este provoque una infección.
La extensa población de anticuerpos y su diversidad se genera por combinaciones al azar de un juego de segmentos genéticos que codifican diferentes lugares de unión al antígeno (o paratopos), que posteriormente sufren mutaciones aleatorias en esta zona del gen del anticuerpo, lo cual origina una diversidad aún mayor.[2][4] Los genes de los anticuerpos también se reorganizan en un proceso conocido como conmutación de clase de inmunoglobulina que cambia la base de la cadena pesada por otra, creando un isotipo de anticuerpo diferente que mantiene la región variable específica para el antígeno diana. Esto posibilita que un solo anticuerpo pueda ser usado por las diferentes partes del sistema inmune. La producción de anticuerpos es la función principal del sistema inmunitario humoral.[5] Los anticuerpos , que son unas proteínas que forman parte del sistema inmune , circulan por sangre. Cuando reconocen sustancias extrañas para el organismo, como los virus y las bacterias o sus toxinas , las neutralizan.
Anticuerpos, inmunoglobulinas y gammaglobulinas
En general, como ya se dijo en la introducción, se considera que anticuerpo e inmunoglobulina son equivalentes, haciendo referencia el primer término a la función, mientras que el segundo alude a la estructura. El término gammaglobulina se debe a las propiedades electroforéticas de las inmunoglobulinas solubles en suero, si bien algunas inmunoglobulinas migran con las fracciones alfa, beta e incluso con la albúmina.
En 1890 comenzó el estudio de los anticuerpos cuando Emil Adolf von Behring y Shibasaburo Kitasato describieron la actividad de los anticuerpos contra la difteria y la toxina tetánica. Behring y Kitasato propusieron la teoría de la inmunidad humoral, que establecía la existencia de un mediador en el suero sanguíneo que podría reaccionar con un antígeno extraño, dándole el nombre de anticuerpo.[6][7] Su idea llevó en 1897 a Paul Ehrlich a proponer la teoría de la cadena lateral de la interacción entre antígeno y anticuerpo y a lanzar la hipótesis de que existían receptores (descritos como "cadenas laterales") en la superficie de las células que se podrían unir específicamente a toxinas —en una interacción de tipo llave-cerradura— y que esta reacción de acoplamiento era el desencadenante de la producción de anticuerpos.[8]
En 1904, siguiendo la idea de otros investigadores de que los anticuerpos se daban libres en la sangre, Almroth Wright sugirió que los anticuerpos solubles revestían las bacterias para señalarlas para su fagocitosis y destrucción en un proceso denominado opsonización.[9]
En los años 1920, Michael Heidelberger y Oswald Avery descubrieron la naturaleza de los postulados anticuerpos al observar que los antígenos podían ser precipitados por ellos y demostrando que éstos eran un tipo de proteínas.[10]

A finales de los años 1930 John Marrack examinó las propiedades bioquímicas de las uniones antígeno-anticuerpo.[11] Luego, en los años 1940 tiene lugar el siguiente avance de importancia, cuando Linus Pauling confirmó la teoría de la llave y la cerradura propuesta por Ehrlich mostrando que las interacciones entre anticuerpos y antígenos dependían más de su forma que de su composición química.[12] En 1948, Astrid Fagreaus descubrió que los linfocitos B en su forma de célula plasmática eran responsables de la producción de anticuerpos.[13]
Los siguientes trabajos de investigación se concentraron en la caracterización de la estructura molecular de los anticuerpos:
- A principios de los años 1960 se produce el principal avance en este sentido, con el descubrimiento por Gerald M. Edelman y Joseph Gally de la cadena ligera,[14] y la comprensión de que esta era idéntica a la proteína de Bence Jones descrita en 1845 por Henry Bence Jones.[15] Edelman continuó con el descubrimiento de que los anticuerpos estaban compuestos por cadenas ligeras y pesadas unidas por enlaces disulfuro.
- Por las mismas fechas, Rodney Porter caracterizó las regiones de unión del anticuerpo (Fab) y la cola del anticuerpo (Fc) en el tipo IgG.[16] Conjuntamente, estos científicos dedujeron la estructura y la secuencia completa de aminoácidos de la IgG, por lo cual recibieron ex aequo el premio Nobel de fisiología y medicina en 1972.[16]
- Mientras la mayoría de estos primeros estudios se fijaron en las IgM e IgG, se identificaron otros isotipos de inmunoglobulina en los años 1960: Thomas Tomasi descubrió los anticuerpos secretados (IgA)[17] y David Rowe y John Fahey identificaron la IgD,[18] y la IgE fue identificada por Kikishige Ishizaka y Teruki Ishizaka como una clase de anticuerpos implicados en reacciones alérgicas.[19]
- En 1975 César Milstein y Georges J. F. Köhler idean el método para la producción de anticuerpos monoclonales.[20] En 1976, los estudios genéticos revelaron la base de la vasta diversidad de los anticuerpos al ser identificada la recombinación somática de los genes de inmunoglobulina por Susumu Tonegawa.[21]
Formas de anticuerpos
Los linfocitos B activados se diferencian en células plasmáticas, cuyo papel es la producción de anticuerpos solubles o bien en linfocitos B de memoria, que sobreviven en el organismo durante los años siguientes para posibilitar que el sistema inmune recuerde el antígeno y responda más rápido a futuras exposiciones al agente inmunógeno.[22] Los anticuerpos son, por tanto, un producto esencial del sistema inmunitario adaptativo que aprenden y recuerdan las respuestas a patógenos invasores. Los anticuerpos se encuentran en dos formas: en forma soluble secretada en la sangre y otros fluidos del cuerpo y en forma unida a la membrana celular que está anclada a la superficie de un linfocito B.
Forma soluble
Los anticuerpos solubles son secretados por un linfocito B activado (en su forma de célula plasmática) para unirse a sustancias extrañas y señalizarlas para su destrucción por el resto del sistema inmune. También se les podría llamar anticuerpos libres hasta que se unen a un antígeno y acaban como parte de un complejo antígeno-anticuerpo o como anticuerpos secretados.
En estas formas solubles se unen a las inmunoglobulinas moléculas adicionales. En la IgM, por ejemplo, encontramos una glucoproteína unida a la Fracción constante mediante puentes disulfuro de unos 15 KD llamada cadena J. Al isotipo IgA, además, se le une la llamada "pieza de secreción". Se trata de una glucoproteína que se forma en las células epiteliales y glándulas exocrinas, y que posteriormente se une a la inmunoglobulina para facilitar su secreción. (Peña, 1998)
Forma anclada a membrana
La forma anclada a membrana de un anticuerpo se podría llamar inmunoglobulina de superficie (sIg) o inmunoglobulina de membrana (mIg), que no es secretado: siempre está asociado a la membrana celular. Forma parte del receptor del linfocito B (BCR), que permite a este detectar cuando un antígeno específico está presente en el organismo, desencadenando la activación del linfocito B.[23] El BCR se compone de anticuerpos IgD o IgM unidos a la superficie de membrana y sus heterodímeros asociados Ig-α e Ig-β que tienen capaz de producir la transducción de señal del reconocimiento del anticuerpo a la célula.[24] Un linfocito B humano típico tiene entre 50 000 y 100 000 anticuerpos unidos a su superficie.[24] Tras el acoplamiento del antígeno, éstos se agrupan en grandes parches cuyo diámetro puede exceder de 1 μm en balsas lipídicas que aíslan los BCR (receptores de la célula B) de la mayor parte de los restantes receptores de señalización celular.[24] Estos parches podrían mejorar la eficiencia de la respuesta inmune celular.[25] En los seres humanos, la superficie celular está libre de otras proteínas alrededor de los receptores de los linfocitos B en distancias de algunos miles de angstroms,[24] lo cual reduce de tal manera las influencias que compiten con su función, que incluso aísla a los BCR.
Isotipos, alotipos e idiotipos
| Nombre | Tipos | Descripción | Complejos de anticuerpos |
| IgA | 2 | Se encuentra en las mucosas, como el tubo digestivo, el tracto respiratorio y el tracto urogenital. Impide su colonización por patógenos.[26] También se encuentran en la saliva, las lágrimas y la leche. | 
|
| IgD | 1 | Su función consiste principalmente en servir de receptor de antígenos en los linfocitos B que no han sido expuestos a los antígenos.[27] Su función está menos definida que en otros isotipos. | |
| IgE | 1 | Se une a alérgeno y desencadena la liberación de histamina de las células cebadas y basófilos y está implicada en la alergia. También protegen contra gusanos parásitos.[5] | |
| IgG | 4 | Proporcionan, en sus cuatro formas, la mayor parte de la protección inmunitaria basada en anticuerpos contra los patógenos invasores.[5] Es el único anticuerpo capaz de cruzar la placenta para proporcionar al feto inmunidad pasiva. | |
| IgM | 1 | Se expresa en la superficie de los linfocitos B y en forma de secreción con gran avidez por su diana. Elimina los patógenos en los estadios tempranos de la respuesta inmune mediada por linfocitos B (humoral) hasta que existen suficientes IgGs.[5][27] |
Los anticuerpos pueden presentarse en distintas variedades conocidas como isotipos o clases. En mamíferos placentados existen cinco isotipos de anticuerpos conocidos como IgA, IgD, IgE, IgG e IgM. Se nombran mediante el prefijo "Ig" que significa inmunoglobulina y difieren en sus propiedades biológicas, localizaciones funcionales y capacidad para reconocer diferentes tipos de antígenos como se muestra en la tabla.[28]
El isotipo cambia durante el desarrollo y la activación de los linfocitos B. Antes de la maduración de estos últimos, cuando aún no se han expuesto a su antígeno, se conocen como linfocitos B vírgenes y solo expresan el isotipo IgM en su forma anclada a la superficie celular. Los linfocitos comienzan a expresar tanto IgM como IgD cuando alcanzan la madurez y en ese momento están listos para responder a su antígeno.[29] La activación de los linfocitos B sigue al encuentro y unión de este con su antígeno, lo que estimula a la célula para que se divide y se diferencie en una célula productora de anticuerpos denominada plasmática. En esta forma activada, los linfocitos B comienzan a secretar anticuerpos en lugar de anclarlos a la membrana. Algunas células hijas de los linfocitos B activados sufren un cambio isotípico, un mecanismo que provoca que la producción de anticuerpos en las formas IgM o IgD se trasmute a los otros tipos, IgE, IgA o IgG, que desempeñan distintos papeles en el sistema inmunitario. Pero también existe un anticuerpo que neutraliza y inactiva a los virus se llama "Anticuerpo Neutralizante".
Alotipos
Se entiende por alotipo las pequeñas diferencias en la secuencia de aminoácidos en la región constante de las cadenas ligeras y pesadas de los anticuerpos producidos por los distintos individuos de una especie, que se heredan de forma mendeliana (Peña, 1998).
En seres humanos se han descrito 3 tipos de determinantes alotípicos:
- En 1956 Grubb y Laurell descubren el sistema Gm en la clase de inmunoglobulinas IgG. Este sistema puso de manifiesto los diversos alotipos de las cadenas pesadas. También permite diferenciar cuatro subclases en estas moléculas: IgG1, IgG2, IgG3 e IgG4 y son determinados genéticamente.[30]
- C. Ropartz y colaboradores descubrieron en 1961 el sistema Km (llamado Inv al principio), localizado en la cadena ligera Kappa. Este alotipo está presente en todas las clases de inmunoglobulina.
- También existe el sistema ISf, situado en la cadena pesada γ1 de la IgG1. La expresión de esta especificidad aumenta con la edad, siendo de un 25 % de los sujetos antes de los 20 años hasta un 60 % después de los 70 años en los caucasoides.
- Los alotipos definidos por el sistema Am se sitúan en las IgA, y más precisamente en las cadenas α2. Existen dos isotipos, α1 y α2, que caracterizan las subclases Am1 y Am2 de las IgA. (Staff, 2003)
Idiotipo
El idiotipo es el epítopo propio de una molécula perteneciente a un clon en particular. Este elemento forma parte o está muy próximo al lugar de reconocimiento del antígeno, y está situado en la porción variable Fab. En otras palabras, es el paratopo, o la región cercana de una inmunoglobulina puede ser reconocido como un epitopo por ciertos linfocitos (Staff, 2003). Según la Teoría de Jerne, La formación de anticuerpos antiidiotipo formaría una red (red de Jerne) cuya función sería la regulación de la síntesis de nuevas inmunoglobulinas. (Peña, 1998).
Estructura
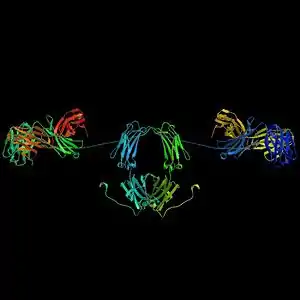
Los anticuerpos son proteínas plasmáticas globulares pesadas (~150 kDa), también conocidas como inmunoglobulinas. Tienen cadenas de azúcares unidas a alguno de sus residuos aminoácido.[31] En otras palabras, los anticuerpos son glucoproteínas. La unidad básica funcional de cada anticuerpo es el monómero de inmunoglobulina, que contiene una sola unidad de Ig. Los anticuerpos secretados también pueden ser diméricos con dos unidades Ig, como en el caso de las IgA, tetraméricos con cuatro unidades Ig como en el caso de las IgM de teleósteo, o pentaméricos con cinco unidades de IgM, como en el caso de las IgM de mamíferos.[32]

Primeros trabajos
Las primeras investigaciones sobre la estructura de los anticuerpos fueron realizados mediante sencillas digestiones con pepsina y papaína por Rodney Robert Porter y Gerald M. Edelman, seguidas de electroforesis. Ambos recibieron por ello el Premio Nobel de medicina en 1972. También fue importante la figura de Alfred Nisonoff:
- En los años 1950, Porter procede a hacer una digestión suave con papaína, obteniendo tres fragmentos, dos de los cuales retenían la especificidad de antígeno (Fab), mientras que el tercero no mostraba actividad de unión, mientras que se podía cristalizar (Fc).
- En 1959, Edelman, utilizando 2-Mercaptoetanol y urea, seguido de electroforesis, consigue aislar las cadenas ligeras y pesadas, al disociar sus enlaces disulfuro y no covalentes.
- Ese mismo año, Porter identifica los componentes de las cadenas ligeras y pesadas que se encontraban en sus fragmentos de papaína y pepsina, y consigue sus pesos moleculares.
- En 1960, Nisonoff demostró que la digestión con pepsina de IgG producía un fragmento bivalente, que en realidad está formado por otros dos, que él denominó F (ab')2.[34]
Dominios de inmunoglobulina
El monómero de Ig es una molécula en forma de "Y" que consta de dos cadenas de polipéptido; dos cadenas pesadas idénticas y dos cadenas ligeras idénticas conectadas por enlaces disulfuro.[28] Cada cadena se compone de dominios estructurales llamados dominios Ig. Estos dominios contienen entre 70 y 110 aminoácidos y se clasifican en diferentes categorías, por ejemplo en variables (IgV) y constantes (IgC) de acuerdo con su tamaño y función.[35] Tienen un "pliegue inmunoglobulina" característico en el cual dos láminas beta generan una forma de "sándwich", permaneciendo juntas por interacciones entre cisteínas bien conservadas a lo largo de la evolución, así como otros aminoácidos cargados.
Cadena pesada
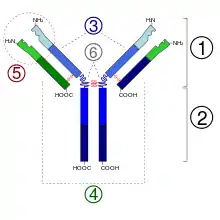
2. Región Fc
3. Cadena pesada con un dominio variable (VH) seguido por un dominio constante (CH1), una región bisagra, y dos más constantes, los dominios (CH2 y CH3).
4. Cadena ligera con un dominio variable (VL) y uno constante (CL)
5. Lugar de unión al antígeno (paratopo)
6. Regiones bisagra.
Hay cinco tipos de Ig en mamíferos que se nombran por letras griegas: α, δ, ε, γ y μ.[3] El tipo de cadena pesada presente define la clase (isotipo) del anticuerpo. Estas cadenas se encuentran en los anticuerpos IgA, IgD, IgE, IgG, e IgM respectivamente. Las distintas cadenas pesadas difieren en tamaño y composición: α y γ contienen aproximadamente 450 aminoácidos, mientras que μ y ε poseen aproximadamente 550 aminoácidos.[3]
Las cadenas pesadas γ, α y δ tienen una región constante compuesta de tres dominios estructurales Ig en tándem y una región bisagra para proporcionarle flexibilidad.[28] Las cadenas pesadas μ y ε tienen una región constante compuesta por cuatro dominios inmunoglobulina.[3] La región variable de la cadena pesada difiere en los anticuerpos producidos en los diferentes linfocitos B, pero es idéntica para todos los anticuerpos producidos por el mismo linfocito B o por su línea clonal. La región variable de cada cadena pesada es de aproximadamente 110 aminoácidos y está compuesto por un único dominio Ig.
Recientemente se ha podido determinar la topología in vivo del gen de la cadena pesada, Igh, siendo este uno de los primeros estudios en este campo. El resultado es que la cromatina se dispone formando giros sucesivos unidos por "linkers", dando lugar a formas similares a una flor. La posición relativa de los distintos segmentos varía drásticamente a lo largo del desarrollo del linfocito B, permitiendo así un mayor rango de interacciones genómicas.[36]
Cadena ligera
En los mamíferos hay dos tipos de cadena ligera, llamados lambda (λ) y kappa (κ).[3] Una cadena ligera contiene dos dominios sucesivos: un dominio constante y un dominio variable. La longitud aproximada de la cadena ligera es de 211 a 217 aminoácidos.[3] Cada anticuerpo contiene dos cadenas ligeras que son siempre idénticas. Solo un tipo de cadena ligera, κ o λ, está presente dentro del mismo anticuerpo en mamíferos. Otros tipos de cadenas ligeras como la cadena iota (ι), se encuentran en los vertebrados inferiores como los condrictios y teleósteos.
Regiones Fab y Fc

Región Fc: fragmento de unión a la célula inmunitaria.
Región Fab
Algunas partes del anticuerpo tienen funciones únicas. Los "extremos de la Y", por ejemplo, contienen el lugar que se une al antígeno y por tanto, reconoce elementos extraños específicos. Esta región del anticuerpo se llama fragmento de unión al antígeno o región Fab. Está compuesta de un dominio constante y otro variable de cada una de las cadenas ligera y pesada del anticuerpo.[37] El paratopo está conformado por los dominios variables de la cadena pesada y ligera en el extremo amino terminal del monómero de anticuerpo.
Región Fc
El papel que desempeña el sector de "la base de la Y", consiste en modular la actividad de la célula inmunitaria. Esta región se llama Fc (de fragmento cristalizable) y está compuesta por dos o tres dominios constantes de ambas cadenas pesadas, dependiendo de la clase del anticuerpo.[3] Mediante la unión a proteínas específicas la región Fc se asegura que cada anticuerpo genera una respuesta inmune apropiada para un antígeno dado.[38] La región Fc también se une a varios receptores celulares como el receptor del Fc y otras moléculas del sistema inmunitario como las proteínas del complemento. Al efectuar esto, media en diferentes efectos fisiológicos incluyendo la opsonización, lisis celular y desgranulación de las células cebadas, basófilos y eosinófilos.[28][39]
Función

Puesto que los anticuerpos se dan de forma libre en el torrente sanguíneo, se dice que son parte del sistema inmunitario humoral. Los anticuerpos circulantes son producidos por líneas clonales de linfocitos B que responden específicamente a un antígeno que puede ser un fragmento de proteína de la cápside viral, por ejemplo. Los anticuerpos contribuyen a la inmunidad de tres formas distintas: pueden impedir que los patógenos entren en las células o las dañen al unirse a ellas (neutralización). Pueden estimular la eliminación de un patógeno por los macrófagos y otras células revistiendo al patógeno (opsonización) y pueden desencadenar la destrucción directa del patógeno estimulando otras respuestas inmunes como la vía del complemento (lisis).[40]
Activación del complemento
Los anticuerpos que se unen a la superficie de los antígenos, por ejemplo, en una bacteria, atraen los primeros componentes de la cascada del complemento mediante su región Fc e inician la activación del sistema "clásico" del complemento.[40] Esto acaba con la muerte de la bacteria de dos formas:[5] Primero, la unión de las moléculas del complemento con el anticuerpo marca al microbio para la ingestión por los fagocitos en un proceso llamado opsonización. Estos fagocitos son atraídos por ciertas moléculas del complemento. En segundo lugar, algunos componentes del sistema del complemento forman un complejo de ataque a membrana para ayudar a los anticuerpos a matar a la bacteria por medio de lisis. Los anticuerpos más efectivos en la activación del Sistema del Complemento son los de tipo IgM y los de tipo IgG subclase 1 y 3 (IgG1 e IgG3).[41]
Activación de células efectoras
Para combatir a los patógenos que se replican en el exterior de las células, los anticuerpos se unen a los patógenos para ensamblarlos juntos provocando su aglutinación. Puesto que un anticuerpo tiene al menos dos paratopos se puede unir a más de un antígeno acoplándose a epítopos idénticos portados en las superficies de esos antígenos. Revistiendo al patógeno, los anticuerpos estimulan las funciones efectoras contra este en las células que reconocen la región Fc.[5]
Aquellas células que reconocen los patógenos revestidos tienen receptores del Fc que, como su nombre indica, interactúan con la región Fc de los anticuerpos IgA, IgG, e IgE. El acoplamiento de un anticuerpo particular con el receptor Fc de una determinada célula desencadena en ella una función efectora: los fagocitos realizarán la fagocitosis, las células cebadas y los neutrófilos producirán la degranulación, las células asesinas naturales liberarán citoquinas y moléculas citotóxicas que finalmente acabarán con la destrucción del microbio invasor. Los receptores Fc son específicos del isotipo, lo que da una mayor flexibilidad al sistema inmune, afectando solo al mecanismo inmune adecuado para los distintos patógenos.[3]

Diversidad de las inmunoglobulinas
Prácticamente todos los microorganismos pueden desencadenar la respuesta de los anticuerpos. El reconocimiento y la erradicación con éxito de tipos muy distintos de estos últimos requiere que los anticuerpos posean una enorme diversidad. Su composición de aminoácidos varía para permitirles interactuar con antígenos muy diferentes.[42] Se ha estimado que los seres humanos generan unos 10 000 millones de anticuerpos diferentes, cada uno de ellos capaz de unirse a un epítopo distinto.[43] Aunque se genera un enorme repertorio de diferentes anticuerpos en un mismo individo, el número de genes disponible para fabricar estas proteínas es limitado. En los vertebrados han evolucionado diferentes mecanismos genéticos complejos para permitir que los linfocitos B generen esta diversidad a partir de un número relativamente pequeño de genes de anticuerpos.[44]
Variabilidad de dominios
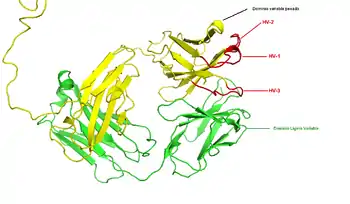
La región (locus) del cromosoma que codifica un anticuerpo es grande y contiene varios genes diferentes para cada dominio del anticuerpo —el locus que contiene los genes para las cadenas pesadas(IGH@) se encuentra en humanos en el cromosoma 14 y los loci que contienen los genes lambda y kappa de la cadena ligera (IGL@ e IGK@) se encuentran en los cromosomas 22 y 2—. Uno de estos dominios es conocido como "dominio variable", que está presente en todas las cadenas ligeras y pesadas de los anticuerpos, pero pueden ser diferentes entre los distintos anticuerpos generados por las variadas líneas de linfocitos B. Las diferencias entre los dominios variables se localizan en tres bucles conocidos como regiones hipervariables (HV-1, HV-2 y HV-3) o regiones determinantes de la complementariedad (CDR1, CDR2 y CDR3). Las CDRs se mantienen entre los dominios variables por regiones de marco conservado. El locus de la cadena pesada contiene unos 65 genes de dominio variable distintos, que difieren en sus CDRs. Combinando estos genes con varios genes de otros dominios se genera un gran contingente de anticuerpos con un alto grado de variabilidad. A esta combinación se la denomina "recombinación V (D) J, que explicamos a continuación.[45]
Recombinación V (D) J
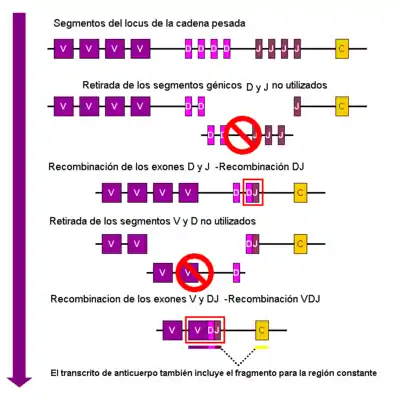
La recombinación somática de las inmunoglobulinas, conocida también como Recombinación V (D) J, consiste en la generación de una región variable de inmunoglobulina exclusiva. La región variable de cada inmunoglobulina pesada está codificada por varias partes, que se conocen como segmentos. Estos son conocidos como segmento variable (V), diversidad (D) y de acoplamiento —joining, en inglés— (J).[44] Los segmentos V, D y J se encuentran en las cadenas pesadas. En las ligeras solo encontramos los segmentos V y J. Hay múltiples copias de todos estos segmentos organizadas en tándem en el genoma de los mamíferos. En la médula ósea cada linfocito B en desarrollo ensambla la región variable de su inmunoglobulina seleccionando y combinando al azar un segmento V con uno D y otro J (o bien uno V y otro J en la cadena ligera). Puesto que existen múltiples copias ligeramente distintas para cada secuencia genética de los segmentos, se darían diferentes combinaciones que mediante este proceso generan un elevado número de paratopos y también diferentes especificidades de antígeno.[2]
Tras la producción de una inmunoglobulina funcional por un linfocito B durante la recombinación V(D)J no podrá expresar ninguna región variable diferente (a este proceso se le conoce como exclusión alélica). Así pues, cada linfocito B solo puede producir anticuerpos que contienen un solo tipo de cadena variable.[3][46]
Hipermutación somática y maduración de la afinidad
Otro mecanismo que genera diversidad en los anticuerpos tiene lugar en los linfocitos B maduros. Tras la activación por antígeno, los linfocitos B comienzan a proliferar rápidamente. En estas células en rápida división, los genes que codifican los dominios variables de las cadenas pesadas y ligeras sufren una gran tasa de mutación puntual mediante un proceso llamado hipermutación somática (SHM). Esta produce aproximadamente el cambio de un nucleótido por gen variable y célula en cada división celular.[4] Como consecuencia, cualquier célula hija de una línea de linfocitos B adquiere una ligera diferencia en la secuencia de aminoácidos de los dominios variables de sus cadenas de anticuerpos.
La hipermutación somática sirve para incrementar la diversidad del reservorio de anticuerpos e influye en la afinidad de la unión entre el antígeno y el anticuerpo.[47] Algunas mutaciones puntuales terminarán por producir anticuerpos que tienen interacciones más débiles (baja afinidad) con su antígeno que el anticuerpo original, mientras que otras generarán anticuerpos con una interacción más fuerte (alta afinidad).[48] Los linfocitos B que expresan anticuerpos de elevada afinidad en su superficie recibirán una fuerte señal para que sobrevivan durante las interacciones con otras células, mientras que las que expresan anticuerpos de baja afinidad morirán por apoptosis.[48] Así pues, los linfocitos B que expresan anticuerpos con una afinidad más elevada por su antígeno competirán con ventaja contra aquellos de menor afinidad en su función y supervivencia. El proceso de generación de anticuerpos con afinidad aumentada progresivamente se llama maduración de la afinidad. La maduración de la afinidad tiene lugar en los linfocitos B maduros tras la recombinación V(D)J y es dependiente del soporte que reciban de los linfocitos T colaboradores.[49]
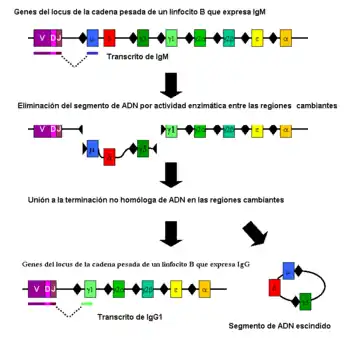
Cambio de clase
La Conmutación de la clase de la inmunoglobulina es un proceso biológico que tiene lugar tras la activación de los linfocitos B, lo cual le permite la producción de diferentes clases de anticuerpos (IgA, IgE, o IgG).[2] Estas clases están definidas por las regiones constantes (C) de la cadena pesada de la inmunoglobulina. Inicialmente, los linfocitos B vírgenes expresan solo IgM e IgD de superficie con regiones de unión al anticuerpo idénticas. Cada isotipo está adaptado para una función distinta y, por tanto, tras la activación, se necesita un anticuerpo con un efector IgG, IgA o IgE para la eliminación eficaz del antígeno. La conmutación de clase permite a la progenie de un solo linfocito B producir anticuerpos de diferentes isotipos. Solo la región constante de la cadena pesada del anticuerpo cambia durante la conmutación de clase. Las regiones variables, y por tanto, la especificidad de antígeno, permanece invariable. De ese modo se producen efectores con la función adecuada para cada amenaza del antígeno. La conmutación de clase se inicia por citoquinas. El isotipo generado depende de que citoquinas estén presentes en el entorno del linfocito B.[50]
El proceso tiene lugar en el gen de la cadena pesada por un mecanismo conocido como recombinación de conmutación de clase ("class switch recombination" o CSR). Este mecanismo se basa en secuencias de nucleótidos conservadas, llamadas regiones de conmutación (Regiones switch o S), que se encuentran en un punto de la secuencia de ADN anterior a los genes de la región constante (excepto en la cadena δ). La hebra de ADN se escinde por la actividad de ciertas enzimas en dos regiones S concretas.[51][52] El exón del dominio variable se vuelve a empalmar mediante un proceso llamado unión de extremos no homóloga ("non-homologous end joining" o NHEJ) a la región constante elegida (γ, α o ε). Este proceso concluye formando un gen de inmunoglobulina que codifica un anticuerpo de un isotipo diferente.[53]
Conversión génica
La conversión génica es un intercambio no recíproco, en el que la secuencia donante no se modifica, mientras que el gen aceptor adquiere un segmento del donante por recombinación homóloga. Aunque este mecanismo para generar diversidad en los anticuerpos se conocía, no se le había dado la suficiente relevancia hasta ahora. Se sabe que es muy importante en aves, las cuales usan en sus cadenas ligeras y pesadas un gran número de pseudogenes semejantes a las secuencias D, situadas al principio de la secuencia del gen de las cadenas de inmunoglobulina. Posteriormente, estos segmentos cambian somáticamente la única región V, pudiendo también estar sometidas a hipermutación.[54] Este mecanismo, curiosamente, también está presente en algunos mamíferos, como los conejos.[55]
Fases finales de la síntesis de inmunoglobulinas
Una vez reagrupados todos los segmentos, se produce un solo mARN, que se poliadenila. Este ARN abandona el núcleo, dirigiéndose a los ribosomas del retículo endoplásmico rugoso, donde comienza su traducción. Posteriormente se produce la glicosilación de los mismos en la parte luminal del RER y el ensamblaje, cuyo proceso es el siguiente H+H → H2+L → H2L2. Constituye una excepción la IgM, uniéndose primero una cadena pesada con una ligera. Su destino final, como receptor o bien ser secretada, depende de si posee o no un fragmento añadido de 19 aminoácidos en la zona C-terminal. Este péptido se incorpora a la síntesis mediante un proceso de splicing. Su presencia determina una región hidrofóbica capaz de anclarse a la membrana celular (Peña, 1998).
Evolución de las inmunoglobulinas
El desarrollo de organismos complejos, con tejidos y varias líneas celulares necesitó del desarrollo de nuevas moléculas para asegurar, por un lado, que las células se adherían a otras de la misma colonia y por otro, la defensa ante posibles intrusos parásitos o patógenos. Tres tipos de moléculas, las lectinas, las LLR y las inmunoglobulinas, han sido utilizadas a lo largo de la evolución en el desarrollo de sistemas inmunitarios. Sus patrones operativos se mezclan en ocasiones para combinar sus propiedades, aunque existen pocas moléculas que contengan los tres, como es el caso del gen de la enfermedad poliquística renal (PKD1).[56]
Muchos estudios aportan pruebas importantes de que la superfamilia de las inmunoglobulinas tienen representantes entre las bacterias y arqueas o que al menos las presentes en este grupo y las de eucariotas podrían tener un antepasado común, desde el cual evolucionaron de forma divergente. Así, se han atribuido a este grupo de proteínas "semejantes a inmunoglobulina" bacterianas (BIg's) al receptor de la Fc de Ig en Streptococcus agalactiae, y la endoglucanasa C de Cellumonas fimi.[57] También existen otros ejemplos como la invasina de Yersinia pseudotuberculosis o las Lig (Leptospiral Ig-like) de diversas especies de Leptospira.[58][59] Tras el hallazgo en Streptococcus se descubrió una proteína de este tipo en el fago T4. En esta ocasión se destacó que su papel estaba relacionado con la adhesividad celular.[60]
Las proteínas con dominios Ig son comunes en eucariotas unicelulares, y hasta cierto punto su estructura es un rasgo conservado.[61] Un ejemplo de ello sería las alfa aglutininas en Saccharomyces cerevisiae. Se trata de moléculas que medían la adhesión celular y que guardan grandes homologías con el grupo CD2-CD4 en humanos, cuyo papel es en parte similar, interviniendo en este último caso la adhesión de los linfocitos T con las células presentadoras de antígenos y las células diana.[62]
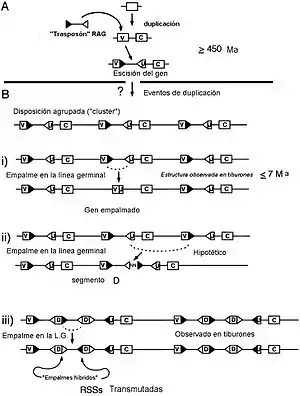
Animales pluricelulares
Sin embargo, es en los grupos de animales pluricelulares más primitivos, los parazoa, donde los científicos intentan hallar respuestas al origen del sistema inmunitario adaptativo.[64] En este sentido, se han dirigido varios trabajos de investigación hacia este grupo, y en especial hacia una esponja considerada como fósil viviente, Geodia cydonium y también Suberites domuncula. En esta primera se encuentran muchos de los tipos de proteínas que también están implicadas en la inmunidad de mamíferos. En especial, hay dos tipos de la superfamilia de las inmunoglobulinas distintas, las unidas a receptor tirosín kinasa, y las moléculas no enzimáticas de adhesión de las esponjas. Curiosamente, los dominios correspondientes ya demuestran polimorfismo, y aún más, aunque cumplen papeles que son simultáneamente de receptores, y de moléculas de adherencia celular, se sobreregulan en experimentos de injerto.[65]
En definitiva, la superfamilia de las inmunoglobulinas intervino en el surgimiento de la multicelularidad al mantener la integridad estructural de los organismos distinguiendo de lo propio de lo ajeno. Esto es debido a que gracias a sus capacidades de generar módulos, de unirse específicamente a otras proteínas y de formar bastones, así como de oligomerizarse y generar diversidad por splicing alternativo a partir de material genético limitado, se convierten en ideales para mediar la adherencia celular y como receptores de superficie de membrana.[66][67]
En la búsqueda de precedentes del sistema inmunitario adaptativo, encontramos varios ejemplos de proteínas de la superfamilia de las Ig en protóstomos que cumplen un papel en la defensa inmunitaria, como la hemolina en gusanos de seda, o la proteína Dscam en Drosophila melanogaster, así como proteínas relacionadas con el fibrinógeno con dominios Ig (FREP) en gasterópodos. Algunas de estas proteínas, que representan una barrera de tipo innato, pueden tener isoformas solubles y ancladas a membrana, y generar diversidad por splicing alternativo, y en zonas de la molécula diferentes a las cadenas variables de vertebrados.[68]
Deuteróstomos
Muchos de los elementos del sistema inmune adaptativo, incluidas las células especializadas, están ya preconfigurados en los organismos más basales de los deuteróstomos. Se han realizado trabajos en el erizo de mar Strongylocentrotus purpuratus, encontrándose un rico sistema inmunitario con homólogos de importantes reguladores inmunitarios y hematopoyéticos de vertebrados, algunos de ellos críticos. Se especula por ello que la presión evolutiva clave para el desarrollo del complejo sistema inmunitario en deuteróstomos no fue tanto la amenaza de patógenos como la existencia de una rica variedad de organismos simbiontes, circunstancia que los propios seres humanos ponemos en evidencia en nuestra flora intestinal.[69]
Como ilustración de este punto, se ha visto que el 60 % de las especies de equinodermos se asocian con simbiontes bacterianos.[70] En tunicados continúa el aumento de la complejidad del sistema inmune. En la ascidia Botryllus schlosseri, durante experimentos de injertos no compatibles, se detectaron muchas proteínas que revelan un complejo sistema inmune innato y algunas proteínas con dominio inmunoglobulina.[71][72] Y lo que resulta más sorprendente, también se puede encontrar un homólogo convincente de RAG1, contiguo a una estructura similar a RAG2. Posteriormente expondremos la importancia de esto último.[73] Sin embargo, es en cefalocordados donde encontramos las primeras huellas de nuestras actuales inmunoglobulinas. Se han realizado múltiples estudios en el anfioxo Branchiostoma floridae, encontrando unas curiosas proteínas, llamadas VCBP (por Proteínas tipo V que contienen dominios que se unen a quitina) con grandes homologías con las regiones V (variables) de las inmunoglobulinas, ciertamente implicadas en la respuesta inmunitaria, pero carentes de su variabilidad. Estudios cristalográficos han demostrado que probablemente se trata de una molécula semejante al ancestro de las actuales regiones variables de vertebrados.[74][75][76]
En los actuales agnatos se dan alguno de los rasgos que identifican un moderno sistema inmunitario adaptativo, mientras que otros están ausentes. Por una parte, existen células que ya contienen gran parte de la maquinaria molecular de los linfocitos. Esto sugiere una evolución de este tipo celular en los vertebrados más basales, y posiblemente en un protocordado. Existen varias proteínas Ig con dominios semejantes a V, que incluso contienen regiones V y J, aunque están codificados en un único exón y no es reorganizable. Sin embargo, no poseen un sistema inmunitario como el de los vertebrados, basado en los clásicos anticuerpos solubles, receptores de membrana, reorganización y empalme por RAG. En lugar de ello, esta función es asumida por una serie de proteínas ricas en repeticiones de leucina, que incluso pueden sufrir una compleja recombinación, a resultas de la cual se obtiene una variabilidad equiparable a la de los anticuerpos (1014). Esto constituye un extraordinario ejemplo de evolución paralela.[77]
Gnatostomados
Todos los autores revisados en este artículo coinciden en que la emergencia del moderno sistema inmunitario tuvo que suceder hace 500 millones de años, durante la explosión cámbrica. Probablemente lo harían dentro de un contexto en el que existirían muchas formas y combinaciones de módulos de proteínas de las que muchas desaparecerían por las presiones selectivas. En este sentido, una de las cuestiones que suscita el apartado anterior es que si la evidencia paleontológica indica que los peces mandibulados actuales proceden de los agnatos, y estos carecen del mismo sistema recombinación de los modernos sistemas inmunitarios, Seguramente debió existir un antepasado común, un ostracodermo ancestral que poseyera ambos sistemas. De acuerdo con este punto de vista, el sistema de recombinación V (D) J probablemente representa un desarrollo evolutivo convergente en una rama de los ostracodermos que precedió a la línea de los gnatóstomos.[78]
En cuanto a las clases de las inmunoglobulinas, en peces encontramos análogos a la clase IgM, así como la IgD, identificada en muchas especies de teleósteos.[79] También existen muchas exclusivas, como las que contienen las cadenas pesadas ζ y τ. Posiblemente son isotipos anteriores a la IgM en la evolución.[80][81] En el caso de los condrictios también encontramos isotipos exclusivos, además de IgM. Se trata de las IgW (IgX o IgNARC) y las IgNAR.[82]
El tipo IgG surge en anfibios y continúa en reptiles, mientras que el tipo IgA aparentemente surge en un antepasado común entre aves y mamíferos. El tipo IgE parece ser exclusivo de mamíferos (Peña, 1998).
Aplicaciones médicas

Diagnóstico de enfermedades
En muchos diagnósticos es común la detección de anticuerpos como prueba de confirmación de la patología. Para ello se realiza una prueba serológica.[86] Como ejemplos, en ensayos bioquímicos para el diagnóstico de enfermedades, se estima el título de anticuerpos contra el virus de Epstein-Barr o contra la enfermedad de Lyme.[87] Si no se encuentran esos anticuerpos significa que la persona no está infectada o que lo estuvo hace mucho tiempo y los linfocitos B que generaban estos anticuerpos se han reducido de forma natural.
En la inmunología clínica se valora por nefelometría (o turbidimetría) los niveles de las distintas clases de inmunoglobulinas para caracterizar el perfil de anticuerpos del paciente.[88] Por ejemplo, una observación en elevación del título de las distintas clases de inmunoglobulina puede ser útil en ocasiones para determinar la causa del daño hepático mediante diagnóstico diferencial. En este sentido, un título elevado de IgA indicaría cirrosis alcohólica; si lo que está elevado son las IgM se sospecha de hepatitis viral y cirrosis biliar primaria, mientras que la IgG está elevada en hepatitis vírica, autoinmune y cirrosis.
Las enfermedades autoinmunes se puede diagnosticar por anticuerpos que se unen a epítopos del propio organismo; muchos de ellos se pueden detectar mediante análisis de sangre. Un ejemplo sería el caso de los anticuerpos dirigidos contra los antígenos de superficie de eritrocitos en la anemia hemolítica mediada por el sistema inmunitario, que se detectan mediante la prueba de Coombs.[89] Esta prueba también se usa para rastrear anticuerpos en la preparación de transfusiones de sangre y también en las mujeres en el periodo prenatal.[89]
En la práctica existen muchos métodos inmunodiagnósticos basados en la detección de complejos antígeno-anticuerpo que se utilizan en el diagnóstico de enfermedades infecciosas, por ejemplo ELISA, inmunofluorescencia, Western blot, inmunodifusión e inmunoelectroforesis.
Tratamientos terapéuticos
La terapia de anticuerpos monoclonales se emplea en el tratamiento de enfermedades como la artritis reumatoide,[90] esclerosis múltiple,[91] psoriasis,[92] y muchas formas de cáncer, incluyendo el linfoma no Hodgkin,[93] cáncer colorrectal, cáncer de cabeza y cuello y cáncer de mama.[94] Algunas inmunodeficiencias, como la agammaglobulinemia ligada al cromosoma X y la hipogammaglobulinemia consisten en una carencia parcial o completa de anticuerpos.[95] Estas enfermedades se tratan a veces induciendo una inmunidad a corto plazo llamada inmunidad pasiva. Esta se adquiere a través de la infusión de anticuerpos "prefabricados" en forma de suero humano o animal, inmunoglobulina intravenosa o anticuerpos monoclonales en el individuo afectado.[96]
Terapia prenatal
Las llamadas Rho (D) Inmunoglobulinas o inmunoglobulilas anti-RhD son específicos del antígeno humano Rhesus D también conocido como factor Rhesus.[97] De estos anticuerpos anti-RhD se conocen varias marcas comerciales, como RhoGAM, BayRHo-D, Gamulin Rh, HypRho-D, y WinRho SDF. El factor Rhesus es un antígeno que se encuentra en los eritrocitos. Los individuos Rhesus-positivo (Rh+) exhiben este anticuerpo en el glucocálix de sus eritrocitos, mientras que los individuos (Rh–) carecen de él.
Durante nacimiento normal, la sangre fetal puede pasar a la madre por traumas en el parto o complicaciones del embarazo. En el caso de incompatibilidad Rh entre la madre y el hijo, la consiguiente mezcla de sangre puede sensibilizar a una madre Rh- contra el antígeno Rh del hijo, haciendo que en los siguientes embarazos corran riesgo de eritroblastosis fetal.[98] Los anti-RhD se administran como parte del tratamiento prenatal para prevenir la sensibilización que pudiera tener lugar para evitarlo. Al tratar a la madre con anticuerpos anti-RhD antes e inmediatamente después del trauma y el parto destruye el antígeno Rh del feto en el cuerpo de la madre. Un tema importante es que esto sucede antes de que el antígeno pueda estimular los linfocitos B maternos que más tarde podrían "recordar" el antígeno Rh generando linfocitos B con memoria. Por tanto, su sistema humoral inmune no fabricará anticuerpos anti-Rh y no atacará los antígenos Rhesus de su bebé actual o futuro.[97]
Aplicaciones en la investigación científica

En investigación, los anticuerpos purificados se usan en muchas aplicaciones. Son muy habituales para identificar y localizar proteínas intra y extracelulares. Los anticuerpos se usan en la citometría de flujo para diferenciar los tipos celulares por las proteínas que expresan; los diferentes tipos celulares expresan también diferentes combinaciones de moléculas del cúmulo de diferenciación (CD) en su superficie y producen diferentes proteínas intracelulares, extracelulares y excretables.[99] También se usan en inmunoprecipitación para separar las proteínas y cualquier cosa que esté unida a ellas (co-inmunoprecipitación) de otras moléculas en un lisado de células,[100] en análisis Western blot para identificar proteínas separadas por electroforesis,[101] y en inmunohistoquímica o inmunofluorescencia para examinar la expresión de proteínas en secciones de tejidos o localizar proteínas en el interior de las células con el auxilio de un microscopio.[99][102] Las proteínas también se pueden detectar y cuantificar con anticuerpos, utilizando técnicas ELISA y ELISPOT.[103][104]
Variantes de anticuerpos en medicina e investigación
En ocasiones se necesita producir anticuerpos específicos. Inyectando un antígeno en un mamífero, como ratón, rata o conejo si se requiere poca cantidad; Cabra, oveja o caballo si se requiere grandes cantidades. La sangre aislada de estos animales contiene anticuerpos policlonales —múltiples anticuerpos que se unen al mismo antígeno— en el suero sanguíneo, al cual se denomina antisuero. También se pueden inyectar antígenos en gallinas y recoger en la yema de huevo los anticuerpos policlonales IgY producidos.[105] Sin embargo, para aplicaciones analíticas es necesaria una mayor especificidad, sobre todo si se trata de detectar moléculas muy pequeñas, así como cuando se usan en aplicaciones terapéuticas en las que se desea bloquear o detectar marcadores muy específicos. Por ello la tecnología de los anticuerpos ha generado algunas variantes, entre las que se destacan:
- Anticuerpos monoclonales
- Si se desea obtener anticuerpos específicos para un único epítopo de un antígeno, se aíslan linfocitos secretores de anticuerpos de un animal y se inmortalizan fusionándolos con una línea celular cancerosa. Las células fusionadas se denominan hibridomas y continuarán creciendo y secretando anticuerpo en el cultivo. Se aíslan las células de hibridoma individuales mediante clonado por dilución para generar clones que produzcan todos el mismo anticuerpo. A estos anticuerpos se les denomina anticuerpos monoclonales.[106]
Los anticuerpos mono y policlonales generados se pueden purificar utilizando proteína A/G o cromatografía de afinidad al antígeno.[107]
- Anticuerpos de cadena sencilla
- Es posible generar artificialmente un anticuerpo que cuente solo con las regiones variables de la cadena ligera y pesada, unidas por un pequeño péptido o un solo aminoácido. En este caso tendremos anticuerpos de cadena sencilla o scFv's. Actualmente se aplican en técnicas como la citometría de flujo o la inmunohistoquímica.[108]
- Abzimas
- La mayoría de los anticuerpos se diferencian de otras proteínas por no presentar catálisis enzimática en su función, por lo que tradicionalmente se consideran proteínas de reconocimiento de superficies moleculares. Sin embargo, en la década de los años 90 del siglo XX y principios del siglo XXI diversos estudios de inmunología encontraron anticuerpos con propiedades catalíticas. Dichos anticuerpos han recibido el nombre de abzimas. Es posible encontrarlas en cantidades bajas en el suero de personas sanas. Un ejemplo de la existencia de las abzimas en el cuerpo humano fue la detección de abzimas contra ADN en la leche materna.[109] Entre algunas otras de estas actividades catalíticas detectadas están las de peptidasas inespecíficas y amilolíticas (degradación de almidón). Por otro lado se ha observado un incremento en el nivel de abzimas en enfermedades de tipo autoinmune. Sin embargo, normalmente se fabrican de forma artificial generando anticuerpos contra el compuesto intermediario de una reacción para la que se desea crear una enzima. En algunas ocasiones podrían tener aplicaciones terapéuticas e industriales.[110][111]
- Nanoanticuerpos
Existen propuestas para la utilización terapéutica de anticuerpos monoclonales de camélido, también llamados nanoanticuerpos. Estos son excepcionales en el reino animal, dado su reducido tamaño, debido a que están compuestos únicamente por dos cadenas pesadas.[112] Tales peculiaridades les permitirían acceder a localizaciones celulares y antígenos inaccesibles para los anticuerpos normales, además de ser posible su administración oral.[113]
- Faboterápicos
Para obtener antídotos contra venenos de picaduras por animales como serpientes o artrópodos, se fabrican antisueros mediante suero crudo o bien altamente enriquecido en inmunoglobulinas. Estos procedimientos producían un gran número de reacciones alérgicas, como anafilaxias o la enfermedad del suero. Para evitarlo, en los años 40 y 50 se realizaron estudios de proteólisis para reducir al mínimo la parte de la molécula implicada en la neutralización del veneno. Finalmente se encontró que el fragmento F (ab’)2, resultante de la digestión con pepsina de los anticuerpos, que carece de las zonas efectoras de la molécula, puede neutralizar igualmente venenos. El profesor Alejandro Alagón Cano propuso para este enfoque terapéutico el nombre de faboterapia, observándose una incidencia mucho menor de reacciones adversas al suero, así como un mejor alcance del compartimento extravascular.[114]
Véase también
Referencias
- Litman, G. W., Rast, J. P., Shamblott, M. J., et al. (1993). «Phylogenetic diversification of immunoglobulin genes and the antibody repertoire». Mol. Biol. Evol. 10 (1): 60-72. PMID 8450761.
- Market, Eleonora; Nina Papavasiliou (2003) «V(D)J Recombination and the Evolution of the Adaptive Immune System» PLoS Biology1(1): e16.doi 10.1371/journal.pbio.0000016
- Janeway, C. A., Jr et al. (2001). Immunobiology. (5th ed. edición). Garland Publishing. ISBN 0-8153-3642-X.
- Diaz, M., Casali, P. (2002). «Somatic immunoglobulin hypermutation». Curr Opin Immunol 14 (2): 235-40. PMID 11869898. doi:10.1016/S0952-7915(02)00327-8.
- Pier, G. B., Lyczak, J. B., Wetzler, L. M. (2004). Immunology, Infection, and Immunity. ASM Press. ISBN 1-55581-246-5.
- «Emil von Behring - Biography». Consultado el 5 de junio de 2007.
- AGN (1931). «The Late Baron Shibasaburo Kitasato». Canadian Medical Association Journal: 206.
- Winau, F., Westphal, O., Winau, R. (2004). «Paul Ehrlich--in search of the magic bullet». Microbes Infect. 6 (8): 786-9. PMID 15207826. doi:10.1016/j.micinf.2004.04.003.
- Silverstein, A. M. (2003). «Cellular versus humoral immunology: a century-long dispute». Nat. Immunol. 4 (5): 425-8. PMID 12719732. doi:10.1038/ni0503-425.
- Van Epps, H. L. (2006). «Michael Heidelberger and the demystification of antibodies». J. Exp. Med. 203 (1): 5. PMID 16523537. doi:10.1084/jem.2031fta.
- Marrack, J. R. (1938). Chemistry of antigens and antibodies (2nd ed. edición). Londres: His Majesty's Stationery Office. OCLC 3220539.
- «The Linus Pauling Papers: How Antibodies and Enzymes Work». Consultado el 5 de junio de 2007.
- Silverstein, A. M. (2004). «Labeled antigens and antibodies: the evolution of magic markers and magic bullets». Nat. Immunol. 5 (12): 1211-7. PMID 15549122. doi:10.1038/ni1140. Archivado desde el original el 16 de junio de 2007.
- Edelman, G. M., Gally, J. A. (1962). «The nature of Bence-Jones proteins. Chemical similarities to polypetide chains of myeloma globulins and normal gamma-globulins». J. Exp. Med. 116: 207-27. PMID 13889153.
- Stevens, F. J., Solomon, A., Schiffer, M. (1991). «Bence Jones proteins: a powerful tool for the fundamental study of protein chemistry and pathophysiology». Biochemistry 30 (28): 6803-5. PMID 2069946. doi:10.1021/bi00242a001.
- Raju, T. N. (1999). «The Nobel chronicles. 1972: Gerald M Edelman (b 1929) and Rodney R Porter (1917-85)». Lancet 354 (9183): 1040. PMID 10501404.
- Tomasi TB (1992). «The discovery of secretory IgA and the mucosal immune system». Immunol. Today 13 (10): 416-8. PMID 1343085.
- Preud'homme, J. L., Petit, I., Barra, A., Morel, F., Lecron, J. C., Lelièvre, E. (2000). «Structural and functional properties of membrane and secreted IgD». Mol. Immunol. 37 (15): 871-87. PMID 11282392. doi:10.1016/S0161-5890(01)00006-2.
- Johansson, S. G. (2006). «The discovery of immunoglobulin E». Allergy and asthma proceedings : the official journal of regional and state allergy societies 27 (2 Suppl 1): S3-6. PMID 16722325.
- Raju, T. N. (Jan. de 2000). «The Nobel chronicles. 1984: Niels Kai Jerne, (1911-94); César Milstein (b 1926); and Georges Jean Franz Köhler (1946-95)». The Lancet 355 (9197): 75. PMID 10615922. doi:10.1016/S0140-6736(05)72025-0.
- Hozumi, N., Tonegawa, S. (1976). «Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions». Proc. Natl. Acad. Sci. U.S.A. 73 (10): 3628-32. PMID 824647.
- Borghesi, L., Milcarek, C. (2006). «From B cell to plasma cell: regulation of V(D)J recombination and antibody secretion». Immunol Res 36 (1-3): 27-32. PMID 17337763. doi:10.1385/IR:36:1:27.
- Parker, D. (1993). «T cell-dependent B cell activation». Annu Rev Immunol 11: 331-60. PMID 8476565. doi:10.1146/annurev.iy.11.040193.001555.
- Wintrobe, Maxwell Myer (2004). Wintrobe's clinical hematology. John G. Greer, John Foerster, John N Lukens, George M Rodgers, Frixos Paraskevas (11 edición). Hagerstwon, MD: Lippincott Williams & Wilkins. pp. 453-456. ISBN 0-7817-3650-1.
- Tolar, P., Sohn, H. W., Pierce, S. K. (February de 2008). «Viewing the antigen-induced initiation of B-cell activation in living cells». Immunol. Rev. 221: 64-76. PMID 18275475. doi:10.1111/j.1600-065X.2008.00583.x.
- Underdown B, Schiff J (1986). «Immunoglobulin A: strategic defense initiative at the mucosal surface». Annu Rev Immunol 4: 389-417. PMID 3518747. doi:10.1146/annurev.iy.04.040186.002133.
- Geisberger R, Lamers M, Achatz G (2006). «The riddle of the dual expression of IgM and IgD». Immunology 118 (4): 429-37. PMID 16895553.
- Woof, J., Burton, D. (2004). «Human antibody-Fc receptor interactions illuminated by crystal structures». Nat Rev Immunol 4 (2): 89-99. PMID 15040582. doi:10.1038/nri1266.
- Goding, J. «Allotypes of IgM and IgD receptors in the mouse: a probe for lymphocyte differentiation». Contemp Top Immunobiol 8: 203-43. PMID 357078.
- Grubb, R., and Laurell, A. B., Acta Path. Microb. Scand., 39, 390 (1956). PMID 13381487
- Mattu, T., Pleass, R., Willis, A., Kilian, M., Wormald, M., Lellouch, A., Rudd, P., Woof, J., Dwek, R. (1998). «The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fc alpha receptor interactions». J Biol Chem 273 (4): 2260-72. PMID 9442070. doi:10.1074/jbc.273.4.2260.
- Roux, K. (1999). «Immunoglobulin structure and function as revealed by electron microscopy». Int Arch Allergy Immunol 120 (2): 85-99. PMID 10545762. doi:10.1159/000024226.
- «Antigen binding sites» (en inglés). Archivado desde el original el 19 de abril de 2007.
- Stevenson, J. R. (18 de agosto). «Immunoglobulin Structure and Function». CAS, Universidad de Miami. Archivado desde el original el 25 de septiembre de 2008. Consultado el 25 de agosto de 2008.
- Barclay, A. (2003). «Membrane proteins with immunoglobulin-like domains--a master superfamily of interaction molecules». Semin Immunol 15 (4): 215-23. PMID 14690046. doi:10.1016/S1044-5323(03)00047-2.
- Murre, C. y otros: (2008). «The 3D structure of the immunoglobulin heavy-chain locus: implications for long-range genomic interactions». Cell 133 (2). PMID 18423198.
- Putnam, F. W., Liu, Y. S., Low, T. L. (1979). «Primary structure of a human IgA1 immunoglobulin. IV. Streptococcal IgA1 protease, digestion, Fab and Fc fragments, and the complete amino acid sequence of the alpha 1 heavy chain». J Biol Chem 254 (8): 2865-74. PMID 107164.
- Huber, R. (1980). «Spatial structure of immunoglobulin molecules». Klin Wochenschr 58 (22): 1217-31. PMID 6780722. doi:10.1007/BF01478928.
- Heyman, B. (1996). «Complement and Fc-receptors in regulation of the antibody response». Immunol Lett 54 (2-3): 195-9. PMID 9052877. doi:10.1016/S0165-2478(96)02672-7.
- Ravetch, J., Bolland, S. (2001). «IgG Fc receptors». Annu Rev Immunol 19: 275-90. PMID 11244038. doi:10.1146/annurev.immunol.19.1.275.
- Rus, H., Cudrici, C., Niculescu, F. (2005). «The role of the complement system in innate immunity». Immunol Res 33 (2): 103-12. PMID 16234578. doi:10.1385/IR:33:2:103.
- Mian, I., Bradwell, A., Olson, A. (1991). «Structure, function and properties of antibody binding sites». J Mol Biol 217 (1): 133-51. PMID 1988675. doi:10.1016/0022-2836(91)90617-F.
- Fanning, L. J., Connor, A. M., Wu, G. E. (1996). «Development of the immunoglobulin repertoire». Clin. Immunol. Immunopathol. 79 (1): 1-14. PMID 8612345.
- Nemazee, D. (2006). «Receptor editing in lymphocyte development and central tolerance». Nat Rev Immunol 6 (10): 728-40. PMID 16998507. doi:10.1038/nri1939.
- Peter Parham. "The Immune System. 2nd ed. Garland Science: New York, 2005. pg.47-62
- Bergman, Y., Cedar, H. (2004). «A stepwise epigenetic process controls immunoglobulin allelic exclusion». Nat Rev Immunol 4 (10): 753-61. PMID 15459667. doi:10.1038/nri1458.
- Honjo, T., Habu, S. (1985). «Origin of immune diversity: genetic variation and selection». Annu Rev Biochem 54: 803-30. PMID 3927822. doi:10.1146/annurev.bi.54.070185.004103.
- Or-Guil, M., Wittenbrink, N., Weiser, A. A., Schuchhardt, J. (2007). «Recirculation of germinal center B cells: a multilevel selection strategy for antibody maturation». Immunol. Rev. 216: 130-41. PMID 17367339. doi:10.1111/j.1600-065X.2007.00507.x.
- Neuberger, M., Ehrenstein, M., Rada, C., Sale, J., Batista, F., Williams, G., Milstein, C. (2000). «Memory in the B-cell compartment: antibody affinity maturation». Philos Trans R Soc Lond B Biol Sci 355 (1395): 357-60. PMID 10794054. doi:10.1098/rstb.2000.0573.
- Stavnezer, J., Amemiya, C. T. (2004). «Evolution of isotype switching». Semin. Immunol. 16 (4): 257-75. PMID 15522624. doi:10.1016/j.smim.2004.08.005.
- Durandy, A. (2003). «Activation-induced cytidine deaminase: a dual role in class-switch recombination and somatic hypermutation». Eur. J. Immunol. 33 (8): 2069-73. PMID 12884279. doi:10.1002/eji.200324133.
- Casali P, Zan H (2004). «Class switching and Myc translocation: how does DNA break?». Nat. Immunol. 5 (11): 1101-3. PMID 15496946. doi:10.1038/ni1104-1101.
- Lieber, M. R., Yu, K., Raghavan, S. C. (2006). «Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations». DNA Repair (Amst.) 5 (9-10): 1234-45. PMID 16793349. doi:10.1016/j.dnarep.2006.05.013.
- Weill, J. C. y otros: (1989). «Somatic hyperconversion diversifies the single VH gene of the chicken with a high incidence in the D region». Cell 59.
- Knight, KL: (1992). «Restricted VH gene usage and generation of antibody diversity in rabbit.». annu. Rev. immunol. 10.
- Litman, G.; Cannon, J. P. y Dishaw, L. J. (noviembre de 2005). «Reconstructing immune phylogeny:new perspectives». Nature 5.
- Bateman, A.; Eddy, S. R. y Chothia, C. (1996). «Members of the immunoglobulin superfamily in bacteria». Protein Science 5 (5). PMID 8880921.
- Dersch, P., Isberg, R. R. (2000). «An immunoglobulin superfamily-like domain unique to the Yersinia pseudotuberculosis invasin protein is required for stimulation of bacterial uptake via integrin receptors.». Infect Immun 68 (5). PMID 10768991.
- Matsunaga, J.; Ko, A. I. y colaboradores: (2005). «Pathogenic Leptospira species express surface-exposed proteins belonging to the bacterial immunoglobulin superfamily». Mol Microbiol 49 (4). PMCID PMC1237129.
- Bateman, A., Eddy, S. R., Mesyanzhinov, V. V. (1997). «A member of the immunoglobulin superfamily in bacteriophage T4». Virus Genes 14 (2). PMID 9237357.
- Wojciechowicz, D., Lu, C. F., Kurjan, J., Lipke, P. N. (1993). «Cell surface anchorage and ligand-binding domains of the Saccharomyces cerevisiae cell adhesion protein alpha-agglutinin, a member of the immunoglobulin superfamily». Mol Cell Biol 13 (4). PMID 8455628.
- Grigorescu A, Chen MH, Zhao H, Kahn PC, Lipke PN (2000). «A CD2-based model of yeast alpha-agglutinin elucidates solution properties and binding characteristics». IUBMB Life 50 (2). PMID 11185954.
- Nick Matzke (28 de abril). «Postulated intermediates in the molecular evolution of the Ig and TCR loci». Journal of Experimental Medicine. Consultado el 22 de agosto de 2008.
|obra=y|publicación=redundantes (ayuda) - Müller, C. I., Blumbach, B., Krasko, A., Schröder, H. C. (2001). «Receptor protein-tyrosine phosphatases: origin of domains (catalytic domain, Ig-related domain, fibronectin type III module) based on the sequence of the sponge Geodia cydonium». Gene 262 (1-2). PMID 11179687.
- Kubrycht, J., Borecký, J., Soucek, P., Jezek, P. (2004). «Sequence similarities of protein kinase substrates and inhibitors with immunoglobulins and model immunoglobulin homologue: cell adhesion molecule from the living fossil sponge Geodia cydonium. Mapping of coherent database similarities and implications for evolution of CDR1 and hypermutation». Folia Microbiol (49). 3 PMID 15259763.
- Brümmendorf, T. y Lemmon, V. (2001). «Immunoglobulin superfamily receptors: cis-interactions, intracellular adapters and alternative splicing regulate adhesion». Current opinion in cell biology 13 (5). doi 10.1016/S0955-0674(00)00259-3.
- Strecker, G y otros: (2004). «Molecular recognition between glyconectins as an adhesion self-assembly pathway to multicellularity». J Biol Chem. 279 (15). PMID 14701844.
- «The Evolution of Adaptative Immune Systems». Cell (124). 2006. DOI 10.1016/j.cell.2006.02.001.
- Litman, GW y otros: (2006). «Genomic Insights into the Immune System of the Sea Urchin». Science 314 (5801). DOI 10.1126/science.1134301.
- Noverr, M. C. y Huffnagle, G. B. «Does the microbiota regulate immune responses outside the gut?». Trends Microbiol 12. PMID.
- Oren, M., Douek, J., Fishelson, Z., Rinkevich, B. (2007). «Identification of immune-relevant genes in histoincompatible rejecting colonies of the tunicate Botryllus schlosseri». Dev Comp Immunol 31 (9). PMID 17287019.
- Pancer, Z., Diehl-Seifert, B., Rinkevich, B., Müller, W. E. (1997). «A novel tunicate (Botryllus schlosseri) putative C-type lectin features an immunoglobulin domain». DNA Cell Biol. 16 (6). PMID 9212174.
- Kapitonov, V. V.; Jurka, J. (2005). «RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons». PLoS Biol. 3.
- Cannon JP, Haire RN, Litman GW: (2002). «Identification of diversified genes that contain immunoglobulin-like variable regions in a protochordate». Nat Immunol 3 (12). PMID 12415263.
- Hernández Prada JA, Haire RN, Allaire M, Jakoncic J, Stojanoff V, Cannon JP, Litman GW, Ostrov DA: (2006). «Ancient evolutionary origin of diversified variable regions demonstrated by crystal structures of an immune-type receptor in amphioxus». Nature immunology 7 (8). PMID.
- Litman GW, Cannon JP, Dishaw LJ, Haire RN, Eason DD, Yoder JA, Prada JH, Ostrov DA: (2007). «Immunoglobulin variable regions in molecules exhibiting characteristics of innate and adaptive immune receptors». Immunol Res. 38 (1-3). PMID 17917037.
- Cooper, M. D. y Alder, M. N. (2006). «The Evolution of Adaptive Immune Systems». Cell 124. DOI 10.1016/j.cell.2006.02.001.
- Janvier, P. (1999). «Catching the first fish». Nature 402. PMID.
- Stein Tore Solem and Jørgen Stenvik. Antibody repertoire development in teleosts--a review with emphasis on salmonids and Gadus morhua L. Developmental & Comparative Immunology, Volume 30, Issues 1-2, Antibody repertoire development, 2006, Pages 57-76.
- Hansen, J. D., E. D. Landis y R. B. Phillips. «Discovery of a unique Ig heavy-chain isotype (IgT) in rainbow trout: Implications for a distinctive B cell developmental pathway in teleost fish.» Proceedings of the National Academy of Sciences U S A. Volume 102, Issue 19, 2005, pp. 6919-24.
- Danilova, N., J. Bussmann, K. Jekosch, L. A. Steiner. The immunoglobulin heavy-chain locus in zebrafish: identification and expression of a previously unknown isotype, immunoglobulin Z. Nature Immunology, Volume 6, Issue 3, 2005, pages 295-302.
- Dooley, H. y M. F. Flajnik. «Antibody repertoire development in cartilaginous fish.» Developmental & Comparative Immunology, Volume 30, Issues 1-2, Antibody repertoire development, 2006, pp. 43-56.
- Padlan, Eduardo (February de 1994). «Anatomy of the antibody molecule». Mol. Immunol. 31 (3): 169-217. PMID 8114766. doi:10.1016/0161-5890(94)90001-9.
- «New Sculpture Portraying Human Antibody as Protective Angel Installed on Scripps Florida Campus». Archivado desde el original el 18 de noviembre de 2010. Consultado el 12 de diciembre de 2008.
- «Protein sculpture inspired by Vitruvian Man». Archivado desde el original el 18 de noviembre de 2010. Consultado el 12 de diciembre de 2008.
- «Animated depictions of how antibodies are used in ELISA assays». Cellular Technology Ltd.—Europe. Archivado desde el original el 9 de mayo de 2007. Consultado el 8 de mayo de 2007.
- «Animated depictions of how antibodies are used in ELISPOT assays». Cellular Technology Ltd.—Europe. Archivado desde el original el 18 de noviembre de 2010. Consultado el 8 de mayo de 2007.
- Stern, P. (2006). «Current possibilities of turbidimetry and nephelometry». Klin Biochem Metab 14 (3): 146-151. Archivado desde el original el 26 de septiembre de 2007.
- Dean, Laura (2005). «Chapter 4: Hemolytic disease of the newborn». Blood Groups and Red Cell Antigens. NCBI Bethesda (MD): National Library of Medicine (US),.
- Feldmann M, Maini R (2001). «Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned?». Annu Rev Immunol 19: 163-96. PMID 11244034. doi:10.1146/annurev.immunol.19.1.163.
- Doggrell S (2003). «Is natalizumab a breakthrough in the treatment of múltiple sclerosis?». Expert Opin Pharmacother 4 (6): 999-1001. PMID 12783595. doi:10.1517/14656566.4.6.999.
- Krueger, G., Langley, R., Leonardi, C., Yeilding, N., Guzzo, C., Wang, Y., Dooley, L., Lebwohl, M. (2007). «A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis». N Engl J Med 356 (6): 580-92. PMID 17287478. doi:10.1056/NEJMoa062382.
- Plosker, G., Figgitt, D. (2003). «Rituximab: a review of its use in non-Hodgkin's lymphoma and chronic lymphocytic leukaemia». Drugs 63 (8): 803-43. PMID 12662126. doi:10.2165/00003495-200363080-00005.
- Vogel, C., Cobleigh, M., Tripathy, D., Gutheil,, J., Harris, L., Fehrenbacher, L., Slamon, D., Murphy, M., Novotny, W., Burchmore, M., Shak, S., Stewart, S. (2001). «First-line Herceptin monotherapy in metastatic breast cancer». Oncology. 61 Suppl 2: 37-42. PMID 11694786. doi:10.1159/000055400.
- LeBien, T. W. (2000). «Fates of human B-cell precursors». Blood 96 (1): 9-23. PMID 10891425. Archivado desde el original el 18 de noviembre de 2010. Consultado el 8 de julio de 2008.
- Ghaffer, A. (26 de marzo de 2006). «Immunization». Immunology - Chapter 14. University of South Carolina School of Medicine. Archivado desde el original el 27 de junio de 2007. Consultado el 6 de junio de 2007.
- Fung Kee Fun,g K., Eason E., Crane, J., Armson, A., De La Ronde, S., Farine, D., Keenan-Lindsay, L., Leduc, L., Reid, G., Aerde, J., Wilson, R., Davies, G., Désilets, V., Summers, A., Wyatt, P., Young, D. (2003). «Prevention of Rh alloimmunization». J Obstet Gynaecol Can 25 (9): 765-73. PMID 12970812.
- Urbaniak, S., Greiss, M. (2000). «RhD haemolytic disease of the fetus and the newborn». Blood Rev 14 (1): 44-61. PMID 10805260. doi:10.1054/blre.1999.0123.
- Brehm-Stecher, B., Johnson, E. (2004). «Single-cell microbiology: tools, technologies, and applications». Microbiol Mol Biol Rev 68 (3): 538-59. PMID 15353569. doi:10.1128/MMBR.68.3.538-559.2004.
- Williams N (2000). «Immunoprecipitation procedures». Methods Cell Biol 62: 449-53. PMID 10503210. doi:10.1016/S0091-679X(08)61549-6.
- Kurien, B., Scofield, R. (2006). «Western blotting». Methods 38 (4): 283-93. PMID 16483794. doi:10.1016/j.ymeth.2005.11.007.
- Scanziani, E. «Immunohistochemical staining of fixed tissues». Methods Mol Biol 104: 133-40. PMID 9711649.
- Reen, D. J. (1994). «Enzyme-linked immunosorbent assay (ELISA)». Methods Mol Biol. 32: 461-6. PMID 7951745.
- Kalyuzhny, A. E. (2005). «Chemistry and biology of the ELISPOT assay». Methods Mol Biol. 302: 15-31. PMID 15937343.
- Tini, M., Jewell, U. R., Camenisch, G., Chilov, D., Gassmann, M. (2002). «Generation and application of chicken egg-yolk antibodies». Comp. Biochem. Physiol., Part a Mol. Integr. Physiol. 131 (3): 569-74. PMID 11867282.
- Cole, S. P., Campling, B. G., Atlaw, T., Kozbor, D., Roder, J. C. (1984). «Human monoclonal antibodies». Mol. Cell. Biochem. 62 (2): 109-20. PMID 6087121.
- Kabir, S. (2002). «Immunoglobulin purification by affinity chromatography using protein A mimetic ligands prepared by combinatorial chemical synthesis». Immunol Invest 31 (3-4): 263-78. PMID 12472184. doi:10.1081/IMM-120016245.
- Lennard, S. (2001). «Standard Protocols for the Construction of scFv Libraries». Springer protocols. DOI 10.1385/1-59259-240-6:059.
- Altria, K. D. (1996). Capillary Electrophoresis Guidebook Principles, Operation, and Applications, página 226. Humana Press. ISBN 1-59259-538-3.
- Blackburn, G. M. y colaboradores: (1996). «Toward antibody-directed "abzyme" prodrug therapy, ADAPT: carbamate prodrug activation by a catalytic antibody and its in vitro application to human tumor cell killing». PNAS 93 (2). Archivado desde el original el 16 de octubre de 2019. Consultado el 19 de agosto de 2008.
- Kobayashi, Shiro; Helmut Ritter, David Kaplan (2006). Enzyme-Catalyzed Synthesis of Polymers, pág. 206. Birkhäuser. ISBN 3-540-29212-8.
- Hamers-Casterman, C., Atarhouch, T., Muyldermans, S., Robinson, G., Hamers, C., Songa, E. B., Bendahman, N., Hamers, R., «Naturally occurring antibodies devoid of light chains.» Nature. 1993 Jun 3;363(6428):446-8
- «Nanobodies herald a new era in cancer therapy». Medical News. mayo de 2004.
- Cano, A. A. «ANTICUERPOS TERAPEUTICOS: EL CASO DE LOS ANTIVENENOS». Sociedad Mexicana de Bioquímica. Archivado desde el original el 3 de diciembre de 2008. Consultado el 25 de agosto de 2008.
{kind=link}
{kind=link}
Bibliografía
- Janeway, C. A.; Staff, V. V. Traducción de Eva Sanz (2003). Inmunobiología: el sistema inmunitario en condiciones de salud y enfermedad. Elsevier España. ISBN 978-84-458-1176-4.
- Peña Martínez, J. (Coordinador) (1998). Inmunología. Pirámide. ISBN 84-368-1213-1. Disponible una versión en línea en http://www.uco.es
Enlaces externos
 Wikimedia Commons alberga una categoría multimedia sobre Anticuerpo.
Wikimedia Commons alberga una categoría multimedia sobre Anticuerpo.- Animaciones que representan cómo anticuerpos se utilizan en las técnicas de ELISPOT y ELISA (inglés)
- Bibliografía anotada sobre evolución del sistema inmunitario en vertebrados (Inglés)
- Nanobodies (material gráfico) Archivado el 1 de abril de 2016 en Wayback Machine.
- Anticuerpo base de datos
- BBC Mundo: Hallan "superanticuerpo" contra todos los virus de gripe
Nota sobre licencia: Algunos de los contenidos del presente artículo han sido traducidos o modificados de Antikörper, antibody y anticorps de las wikipedias alemana, inglesa y francesa respectivamente, todas ellas bajo licencia GFDL.
| Control de autoridades |
|
|---|
 Datos: Q79460
Datos: Q79460- Multimedia: Antibodies / Q79460
 Recursos didácticos: Anticuerpos
Recursos didácticos: Anticuerpos