Prion
Un prion[1][2] es una proteína mal plegada capaz de transmitir su forma mal plegada a otras variedades de la misma proteína.[3] Produce las encefalopatías espongiformes transmisibles, que son un grupo de enfermedades neurológicas degenerativas tales como la tembladera, la enfermedad de Creutzfeldt-Jakob y la encefalopatía espongiforme bovina.[4][5] Los priones son considerados agentes infecciosos[3] y su forma intracelular puede no contener ácido nucleico.

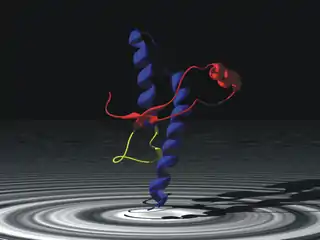
La proteína priónica (abreviada como PrNp) es una glicoproteína presente de forma natural en muchas células (se denomina PrPc), pero puede convertirse en patogénica (PrPSc) como consecuencia de la alteración de su estructura secundaria, lo que conduce a un incorrecto plegamiento de su estructura terciaria.[6] A diferencia del resto de los agentes infecciosos (hongos, bacterias, virus, viroides, etc.), que contienen ácidos nucleicos (ya sea ADN, ARN, o ambos), un prion solamente está compuesto por aminoácidos y no presenta material genético. En comparación con los otros agentes infecciosos mencionados que son autónomos de su huésped, los priones se originan de la mala síntesis proteica del gen PRNP. Sin embargo, se desconoce por qué se pueden transmitir entre individuos, ya que las enfermedades causadas por priones podrían considerarse un tipo de enfermedad genética.
El prion, palabra acuñada en 1982 por Stanley B. Prusiner al investigar una serie de enfermedades de carácter crónico e irreversibles que afectaban al sistema nervioso central,[7] es un acrónimo inglés derivado de las palabras proteína e infección. Los priones son los responsables de las encefalopatías espongiformes transmisibles en una variedad de mamíferos, incluida la encefalopatía espongiforme bovina (EEB, también conocida como «enfermedad de las vacas locas») en el ganado y la enfermedad de Creutzfeldt-Jakob (ECJ) en humanos.[8] Dichas proteínas mutadas forman agregados supramoleculares y son patógenas con plegamientos anómalos ricos en láminas beta, y autorreproducibles.
Los priones se propagan mediante la transmisión de proteínas anómalas con mal plegamiento. Cuando un prion entra en un organismo sano, actúa sobre la forma normal del mismo tipo de proteína existente en el organismo, modificándola y convirtiéndola en prion. Estos priones recién formados pueden pasar a convertir más proteínas, provocando una reacción en cadena que produce grandes cantidades de la proteína prion. Todos los priones conocidos inducen la formación de amiloides plegado, en los que actúan polimerasas formando un agregado que consiste en apretadas hojas β. El período de incubación de las enfermedades prionicas se determina por la tasa de crecimiento exponencial asociados con la replicación de priones, que es un equilibrio entre el crecimiento lineal y la rotura de los agregados (hay que tener en cuenta que la propagación del prion depende de la presencia de la proteína normalmente plegada en la que los priones pueden inducir plegamiento. Los organismos que no expresan la forma normal de la proteína prionica no pueden desarrollar o transmitir la enfermedad).[9]
Función de los priones en estado no patógeno (PrPc)

A pesar de que actualmente hay muy poca información sobre la función que tiene la proteína PrPc (estado no patógeno), algunos experimentos han demostrado que tienen un papel activo en el correcto desarrollo neuronal, que es una proteína capaz de unir específicamente Cu2+ (procesos rédox), y también se han relacionado los priones con proteínas de transducción de señales, la adhesión celular y la regulación y distribución de los receptores de acetilcolina.[10]
Historia
Las primeras referencias a las enfermedades espongiformes transmisibles se remontan al siglo XVIII, cuando varios ganaderos europeos describieron una enfermedad neurodegenerativa letal que afectaba a las ovejas y a las cabras, mal al que se denominó «tembladera». El cerebro de estos animales presentaba un aspecto de esponja, de donde proviene el término «espongiforme». A principios del siglo XX se describieron los primeros casos de encefalopatía espongiforme en el ser humano, y la enfermedad se bautizó con el nombre de enfermedad de Creutzfeldt-Jakob. Posteriormente se demostró que estas enfermedades eran transmisibles. El agente patógeno, el prion, fue descubierto en 1982 por Stanley Prusiner, quien demostró que se trataba de partículas puramente proteicas sin ácido nucleico. En 1997 le fue otorgado el Premio Nobel de Fisiología o Medicina.
Prusiner sometió los priones a distintos tratamientos para alterar las proteínas o los ácidos nucleicos, intentando alterar su capacidad infecciosa. Observó que perdían infectividad si se trataban con fenol (agentes desnaturalizantes de las proteínas, pero no de los ácidos nucleicos), aunque eran resistentes a algunos de los procesos de degradación proteica (como las enzimas proteasas). Sin embargo, si los sometía a la acción de enzimas que atacaban específicamente a los ácidos nucleicos (nucleasas para ADN y ARN), a radiación UV o a la modificación con hidroxilamina, las partículas no perdían su infectividad. Estos estudios indicaron que los priones eran partículas patógenas de naturaleza proteica y sin ácido nucleico. Prusiner consiguió, más adelante, infectar con el prion de la tembladera PrPSc (que causa el prurito lumbar en ovejas) a ratones, consiguiendo un modo para reproducir, obtener y estudiar los priones posteriormente y más a fondo.
Clasificación
Los priones se han detectado en los mamíferos y en las levaduras. Los priones de levaduras no se consideran patógenos. Se han descrito las siguientes proteínas prionicas:
Características de los priones
Una vez obtenido el prion responsable de la patología, comenzó a dilucidarse su estructura. Se trata de una glucoproteína de 27-30 kD, que tiene la misma estructura primaria que una proteína similar presente en el cerebro de la oveja. La modificación estructural, no genética, se debe a un proceso de postraducción. Esto se presenta en funciones muy especiales.
El gen codificando del prion PrPc (gen PRNP) se localiza en el brazo corto del cromosoma 20, no tiene intrones y es un gen autosómico dominante. Se expresa en el tejido neuronal, cardíaco, muscular, pancreático y hepático.
El plegamiento erróneo de la PrPc a PrPSc confiere a la PrPSc dos propiedades que la diferencian de la PrPc: la resistencia parcial a la digestión por proteasas y su insolubilidad. Estas dos propiedades hacen que la PrPSc sea estable y la capacitan para poder formar agregados proteicos responsables de la acumulación de PrPSc en forma de placas amiloides en el tejido nervioso.[13] Las principales diferencias entre la forma normal del prion, PrPc, y la forma patógena, PrPSc son las siguientes:
| PrPc | PrPSc |
|---|---|
| Estructura helicoidal α (4 regiones de proteína globular) | Estructura laminar β (proteína plana) |
| Susceptible a proteasas | Resistente a proteasas |
| Proteína monomérica | Agregados proteicos |
| Monómeros estables | Monómeros poco estables (agregados amiloides) |
| Resistencia normal | Resistencia extrema a la radiación y disolventes fuertes |
| Soluble en detergentes | Insoluble en detergentes |
Es importante destacar que tanto las PrPc como las PrPSc tienen la misma secuencia de aminoácidos, pues derivan del mismo gen.
Contagio de la estructura en lámina beta y formación de placas amiloides
Una proteína globular en forma de hélice alfa de una membrana neuronal entra en contacto con una proteína PrPSc, que actúa de agente infeccioso haciendo que la PrPc adquiera estructura plana en forma de lámina beta, es decir, pasará a ser una PrPSc.[14] Como consecuencia de este cambio conformacional, la nueva proteína no puede ser degradada y actúa como agente infeccioso sobre otros PrPc provocando el mal plegamiento de una manera exponencial. Este hecho es el causante de la acumulación de agregados de PrPSc en forma de placas amiloides, agregados proteicos patógenos que se acumulan en forma de fibras insolubles y que matan las neuronas produciendo agujeros en el cerebro.
Mecanismo de contagio
No se conocen con exactitud los mecanismos a través de los cuales la PrPc pasa a ser PrPSc. Una de las hipótesis es la creación de radicales -SH, cambiando unos aminoácidos presentes comúnmente en las hélices alfa (encontradas en alto porcentaje en las PrPc) por otros, normalmente entre cisteínas que pueden formar puentes disulfuro, hecho que da lugar a un cambio conformacional y dando lugar a un aumento en la proporción de láminas beta.
Según las hipótesis actuales, los aminoácidos que actúan en el cambio conformacional son la metionina, por su capacidad de crear enlaces disulfuro por su radical –SH, así como la cisteína; y la valina, por su proximidad atómica a la Met y Cys.
En el laboratorio se pueden romper los puentes disulfuro mediante dos mecanismos: a partir de la oxidación con ácido perfórmico o mediante la rotura con beta-mercaptoetanol y posterior acetilación con iodoacetato.
Otras hipótesis señalan que la sustitución del aminoácido leucina por prolina puede ser el posible responsable de la desestabilización de la hélice alfa de la proteína y su transformación en agente patógeno, ya que la presencia de prolina impide la formación de esta estructura secundaria; de tal modo que el porcentaje de hélice alfa disminuye de las PrPc a las PrPSc y aumenta considerablemente el de lámina beta.
Transporte hacia el cerebro
A partir de una ingesta contaminada, el agente patógeno es transportado por el epitelio intestinal, desde donde entra a las células M (especializadas en el transporte de macromoléculas y partículas a través de las células del epitelio intestinal) mediante transcitosis. A continuación, el agente entra dentro de las células migratorias y de los macrófagos (sistema inmunitario). Una vez reconocido, se sintetiza un anticuerpo contra el prion, pero no tiene ninguna eficacia, causa por la cual el prion es transportado por el sistema inmunitario y se acumula en el bazo y los ganglios linfáticos, que están muy inervados. Este hecho produce el contagio al tejido nervioso y la consecuente muerte neuronal y por ello, el cerebro adquiere un aspecto esponjoso.[15]
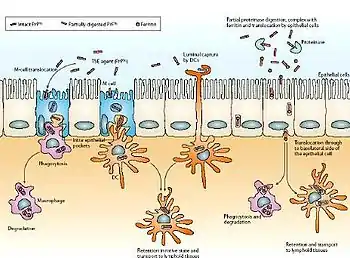
La muerte neuronal se produce porque las PrPSc son insolubles y resistentes a las proteasas de los lisosomas. Este hecho explica la acumulación de PrPSc en los lisosomas, produciendo un aumento de volumen y la consecuente lisis de los lisosomas que acidifican la célula produciendo su muerte.
Enfermedades prionicas
Hay tres formas de enfermedades producidas por priones: formas esporádicas de la enfermedad, que aparecen sin causa aparente y actualmente no tienen explicación científica; formas infecciosas, que son consecuencia de la interacción de la PrPSc sobre la PrPc, que provoca su transformación en PrPSc y formas hereditarias provocadas por alteraciones genéticas que facilitan el plegamiento erróneo de la PrPc.
En los animales

En los animales, las enfermedades asociadas a los priones originan descoordinación en los movimientos, ceguera o la muerte. Las más conocidas son la Encefalopatía Espongiforme Bovina (enfermedad de las vacas locas, registrada por primera vez en el año 1984), causada por la alimentación suplementaria del ganado vacuno con restos ovinos y caprinos (que ya presentaban la enfermedad). Otra enfermedad manifestada en animales es la tembladera (en ovejas y cabras), que no se transmite a los seres humanos, pero sí a los vacunos, capaces de transmitir la enfermedad a los seres humanos. Esta patología también existe en felinos a pesar de que hay muy pocos casos registrados.
En el ser humano
Es interesante destacar que para las enfermedades prionicas humanas existe una sensibilidad genética para ciertos homocigotos en el Codón 129 del gen, los homocigotos Metionina y también Valina tienen más susceptibilidad a padecer la enfermedad.
- Enfermedad de Creutzfeldt-Jakob (ECJ). Es la forma más frecuente; habitualmente se presenta de forma esporádica, que representa un 90 % de los casos registrados de la enfermedad (un caso por cada millón de habitantes, aproximadamente) a partir de los 50-60 años de edad. Un 1 % se considera de origen infeccioso debido a prácticas médicas, se han registrado casos de infección por vía serológica (transfusión sanguínea) y, antiguamente, por el empleo terapéutico de hormonas hipofisarias derivadas de animales o de cadáveres humanos infectados, así como ciertos injertos de duramadre, trasplantes de córnea. Pero únicamente en un 10-15 por ciento de los casos el origen es genético. Se han registrado alrededor de 20-30 mutaciones diversas del gen PRNP. La muerte sobreviene alrededor de los 4-6 meses a partir del diagnóstico.
- Insomnio familiar fatal. Trastorno del sueño habitualmente de origen genético, producido por una mutación N178D en la secuencia del PrP. Se conocen 40 casos hereditarios[16] y un escaso número sin causa genética.
- Nueva variante de la enfermedad de Creutzfeldt-Jakob. Transmitida. Se inició en Gran Bretaña en los años 90 (en el 1996 se publicaron los primeros casos) y se ha relacionado con la ingesta de productos procedentes de reses afectadas de la que, por esa razón, se denomina encefalopatía espongiforme bovina La edad de comienzo es significativamente menor que ECJ, siendo la media de edad de 27 años, duración de la enfermedad es de aproximadamente 14 meses, produciendo la muerte del individuo.
- Enfermedad de Gerstmann-Straüssler-Scheinker. De origen genético.
- Kuru. Transmitida. Restringida a poblaciones de Papúa Nueva Guinea y relacionada con prácticas caníbales. Se considera una enfermedad en extinción. Presenta un periodo de incubación muy variable que oscila desde los 4 años hasta los 40 años. Está relacionado con el consumo de cerebros humanos infectados como acto ritual.
- Encefalopatía espongiforme familiar asociada a una nueva mutación en el gen PrP. Individualizada en una sola familia brasileña, es hereditaria y autosómica dominante.
por estás enfermedades los priones reciben el apodo de "proteínas Zombies"
En especies animales
- «Tembladera» (prurito lumbar) en ovejas
- Encefalopatía espongiforme bovina (llamada enfermedad de las vacas locas)
- Otras (enfermedad caquectizante de alces, encefalopatía espongiforme felina, etc.)
En los microorganismos
También hay microorganismos que se ven afectados por priones. Es el caso del fenotipo PSI en la levadura de cerveza (Saccharomyces cerevisiae).
Los priones en la vida normal de los organismos
Las investigaciones tendentes a describir la naturaleza de los priones y los agregados amioloides que forman permitieron observar proteínas-priones en organismos en estado natural, es decir, de una manera tal que no puede decirse que estén relacionados con una enfermedad. Modelos en organismos del reino de los hongos (Fungi), particularmente en Saccharomyces cerevisiae, han permitido observar las funciones que podrían tener los priones en la vida normal de las células. En estos organismos, los priones desempeñan funciones tales como la regulación metabólica del nitrógeno. Los priones actúan también como mecanismos de la herencia de fenotipos, en el papel de capacitadores evolutivos, y aumentando la diversidad genética, al introducir regiones nuevas en los extremos del genoma.
En el año 2003, E. R. Kandel y sus colaboradores lograron identificar una proteína similar a un prion en Aplysia. Esta proteína, la CPEB, está relacionada con procesos de traducción del ARNm específico en las sinapsis durante los procesos de plasticidad sináptica y de formación de la memoria. La capacidad de guardar información conformacional de los priones los convierte en candidatos para participar en procesos celulares que requieren estabilidad durante periodos prolongados y, dado que los priones son menos susceptibles a la digestión enzimática, es posible que ellos sean mecanismos celulares.
Es probable que los priones participen en procesos como la formación de la memoria a largo plazo, la memoria inmunitaria y la evolución del genoma de muchos organismos. De hecho, con el uso de ratones knockout, ha visto que podrían ser los responsables de evitar la excitación excesiva de los canales de NMDA de las neuronas.
Experimentos en el laboratorio
A lo largo de los años se han llevado a cabo varios experimentos en el laboratorio, mayoritariamente con ratones homocigotos respecto a la falta del gen PRNP (que codifica para la isoforma normal del prion). Se ha observado que los ratones son normales hasta las 70 semanas de vida, momento a partir del cual se registra una importante pérdida de coordinación, temblores al andar e incapacidad de mantener una trayectoria. A las 90 semanas son incapaces de mantenerse, tienen movimientos espásmicos en las extremidades posteriores y muestran la columna vertebral arqueada.
No obstante, este estudio presenta problemáticas, puesto que el periodo de incubación o tiempo de latencia del prion es de gran duración (hasta 4 o 5 años).
Posteriormente se han creado clones de ratón sin el gen PRNP que han crecido y se han desarrollado de una manera normal, con algunas excepciones de ataxia y alteración del ritmo cardíaco más allá de los dos años de edad. En los ratones sin el gen PRNP se ha observado la disminución del cerebelo (encargado de la coordinación) de hasta un tercio, deficiencias neuronales a largo periodo, y ausencia de la enfermedad (puesto que no hay ninguna PrPc a infectar).
Detección de la PrPSc
Mediante electroforesis en geles de poliacrilamida, posterior transferencia a membrana de nitrocelulosa e inmunodetección se puede detectar la presencia de la PrPSc, puesto que es resistente a la proteasa K a diferencia de la PrPc, que no es resistente.
Tratamientos preventivos
Un posible tratamiento para suprimir la acción de la PrPSc sería mediante intervenciones que estabilizaran las supuestas hélices alfa y de esta forma inhibir su conversión a láminas beta, con fármacos que se unan a las cuatro regiones que tiene la hélice alfa de la PrPc.[17] Otra posibilidad es tener en cuenta que los animales que carecen del gen PRNP se desarrollan y tienen una fisiología normal. Partiendo de esta idea, se podría eliminar los genes del cerebro de los enfermos o sintetizar fármacos que bloquearan su expresión, puesto que para que los priones puedan desarrollarse es necesaria la existencia de una reserva de PrPc. El uso de fármacos que inhiban la interacción PrPc-PrPSc o cualquier producto que de alguna manera interfiera en los procesos de endocitosis, exocitosis, tráfico intracelular y degradación proteica, en particular de la PrPc, es otra opción.
Véase también
Referencias
- Real Academia Española. «prion». Diccionario de la lengua española (23.ª edición). Consultado el 3 de junio de 2017.
- «La doble grafía —con tilde o sin ella— que admitía para estas palabras la Ortografía de 1999 (crie o crié, guion o guion, Ruan o Ruán, etc.) no es asimilable a la que presentan las voces con doble acentuación prosódica (v § 2.3.3). En los casos de doble acentuación prosódica, la duplicación de grafías está plenamente justificada porque responde a la posibilidad de que el acento recaiga en dos vocales distintas; así, en la forma esdrújula alvéolo [al.bé.o.lo] es tónica la e, mientras que en la variante llana alveolo [al.be.ó.lo] es tónica la primera o. En cambio, la duplicidad acentual en casos como el de guion/guion, truhan/truhán y similares no se justifica por un cambio de la vocal tónica, que sigue siendo la misma en ambas formas, sino que responde a la consideración de monosílabas o bisílabas de estas palabras según se articule como diptongo o como hiato la secuencia vocálica que contienen: crie [krié], guion [gión], truhan [truán] frente a crié [kri.é], guion [gi.ón], truhán [tru.án]. […] La convención que establece qué secuencias vocálicas se consideran diptongos, triptongos o hiatos a efectos ortográficos debe aplicarse sin excepciones y, en consecuencia, las palabras antes mencionadas se escribirán obligatoriamente sin tilde, sin que resulten admisibles, como establecía la Ortografía de 1999, las grafías con tilde.». Citado en RAE y ASALE (2010). «§ 3.4.2.1.1 Diptongos ortográficos». Ortografía de la lengua española. Madrid: Espasa Calpe. p. 236. ISBN 978-6-070-70653-0. Consultado el 3 de septiembre de 2017.
- «Prion diseases». Diseases and conditions. National Institute of Health.
- Sakudo, A.; Ano, Y.; Onodera, T.; Nitta, K.; Shintani, H.; Ikuta, K.; Tanaka, Y. (2011). «Fundamentals of prions and their inactivation (Review)». International Journal of Molecular Medicine (en inglés) 27 (4): 483-489. ISSN 1107-3756. doi:10.3892/ijmm.2011.605. Consultado el 11 de diciembre de 2013.
- González T, Rubio; Jarque M, Verdecia (2009). «Enfermedades prionicas». MEDISAN (en inglés) 13 (1). Archivado desde el original el 11 de marzo de 2018. Consultado el 12 de diciembre de 2013.
- Ryan, K. J., Ray, C. G., et al (2004). Sherris Medical Microbiology (4th edición). McGraw Hill. pp. 624-8. ISBN 0-8385-8529-9.
- «Stanley B. Prusiner - Autobiography». NobelPrize.org. Consultado el 2 de enero de 2007.
- Prusiner SB (noviembre de 1998). «Prions». Proceedings of the National Academy of Sciences of the United States of America 95 (23): 13363-83. PMC 33918. PMID 9811807. doi:10.1073/pnas.95.23.13363. Consultado el 28 de febrero de 2010.
- Masel J., Jansen, V. A. A., Nowak, M. A. (marzo de 1999). «Quantifying the kinetic parameters of prion replication». Biophysical Chemistry 77 (2-3): 139.152. PMID 10326247. doi:10.1016/S0301-4622(99)00016-2.
- Vertebrate prions subviral agents Comité Internacional de Taxonomía de Virus.
- Fungal prions subviral agents Comité Internacional de Taxonomía de Virus.
- MATHEWS, Christopher K., VAN HOLDE, K. F., AHERN, Kevin G. Bioquímica 2002, Ed. Pearson
- «Copia archivada». Archivado desde el original el 15 de junio de 2011. Consultado el 10 de diciembre de 2010.
- Unidad de sueño - Hospital Universitario Dr. Peset de Valencia
Enlaces externos
 Wikimedia Commons alberga una categoría multimedia sobre Prion.
Wikimedia Commons alberga una categoría multimedia sobre Prion. Wikispecies tiene un artículo sobre Prion.
Wikispecies tiene un artículo sobre Prion.- "Destructores de cerebros", artículo informativo y didáctico sobre las enfermedades producidas por priones.
- Microbiología clínica (En línea). Tema 2: Enfermedades causadas por priones.