Tríada catalítica
El término tríada catalítica se refiere a los tres residuos de aminoácidos que funcionan en conjunto en el centro del sitio activo de algunas enzimas hidrolasas y transferasas (por ejemplo, proteasas, amidasas, esterasas, acilasas, lipasas y β-lactamasas). Una tríada Ácido-Base-Nucleófilo es un motivo común para la generación de un residuo nucleofílico para la catálisis covalente.[1][2] Los residuos aminoacídicos forman una red de relevamiento de cargas que polarizan y activan al nucleófilo, el cual ataca al sustrato, formando un intermediario covalente el cual es luego hidrolizado para regenerar la enzima libre. El residuo nucleofílico es por lo general una serina o una cisteína pero algunas veces puede ser una treonina.
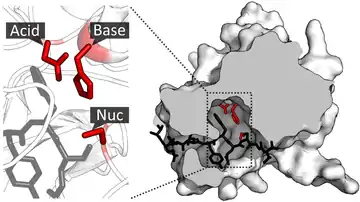
Debido a que las enzimas se pliegan en complejas estructuras tridimensionales, los residuos de la tríada catalítica pueden estar muy alejados unos de otros sobre la secuencia primaria, sin embargo, se encuentran estrechamente relacionados en el plegamiento final.
Además de ejemplificar a la perfección la evolución divergente de función (incluso con el nucleófilo de la tríada), las tríadas catalíticas muestran algunos de los mejores ejemplos de evolución convergente. Las limitaciones químicas del proceso catalítico han conducido a la misma solución catalítica para al menos 23 superfamilias diferentes de enzimas que evolucionaron en forma independiente.[2] Su mecanismo enzimático es por lo tanto uno de los mejor estudiados en toda la bioquímica.[3][4]
Historia
Las estructuras de la tripsina y quimotripsina fueron resueltas por primera vez en los años 1930.[5] El miembro nucleofílico de la tríada de la tripsina y quimotripsina fue ientificado como la serina por modificación con diisopropil fluorofosfato en los años 1950.[6] A estas se le sumaron otras secuencias de proteasas en los años 1960, revelando una familia de proteasas emparentadas,[7][8][9] que actualmente se conoce como la familia S1. En forma simultánea se encontró que las estructuras de la papaína y subtilisina, unas enzimas sin ninguna relación evolutiva con las anteriores, contenían tríadas análogas. El mecanismo de "relevamiento de carga" para la activación del nucleófilo por otros miembros de la tríada se propuso por primera vez a finales de los años 1960.[10] A medida que se iban resolviendo nuevas estructuras enzimáticas por cristalografía de rayos X en los años 1970 y 1980; se fueron encontrando nuevas homologías biológicas y tríadas análogas (como las de la papaína).[11][12][13] El sistema de clasificación MEROPS en las décadas de 1990 y 2010 comenzó a clasificar a las proteasas en superfamilias de enzimas estructuralmente realacionadas, actuando por lo tanto como una base de datos para el registro de la evolución convergente de más de 20 superfamilias.[14][15] La comprensión de las limitaciones dadas por la química a la evolución, que ha conducido a la convergencia de muchas familias de enzimas hasta contener la misma geometría de tríada catalítica, comenzó a ser entendida en la década de 2010.[2] El masivo cuerpo de trabajo en relevamiento de cargas y catálisis covalente dado por tríadas catalíticas ha conducido a que este mecanismo sea uno de los mejor caracterizados de toda la bioquímica.[3][4]
La identidad de los miembros de las tríadas

Nucleófilo
La cadena lateral del residuo nucleofílico lleva a cabo un proceso de catálisis covalente sobre el sustrato. El par solitario de electrones presente en el oxígeno o el azufre del nucleófilo ataca al carbono del grupo carbonilo. Los 20 aminoácidos canónicos no contienen grupos funcionales lo suficientemente nucleofílicos para llevar a cabo muchas reacciones catalíticas particularmente dificultosas. En la naturaleza, los nucleófilos más comúnmente utilizados son el grupo alcohol (OH) de una serina y el grupo tiol/tiolato (SH/S−
) de una cisteína. La introducción del nucleófilo dentro de una tríada consigue hacerlo más activo desde el punto de vista catalítico. Un par de proteasas emplean el grupo alcohol secundario de una treonina, sin embargo, debido al grupo metilo extra, estas proteasas hacen uso de la amida N-terminal como la base de la tríada, en lugar de un aminoácido separado.[1][16]
Base
Ya que no existen aminoácidos naturales que sean nucleófilos fuertes, la base de la tríada catalítica polariza y desprotona al nucleófilo para aumentar su reactividad. Adicionalmente, protona al primer producto y ayuda a la liberación del grupo saliente. Por lo general se trata de una histidina ya que su pKa permite tanto una catálisis básica como un enlace de hidrógeno tanto para el residuo ácido como para desprotonar al residuo nucleofílico. Las β-lactamasas tales como la TEM-1 hacen uso de un residuo de lisina como base. Debido a que el pKa de la lisina es tan alto (pKa=11), un glutamato y varios otros residuos actúan como ácidos para estabilizar su estado desprotonado durante el ciclo catalítico.[17][18] Para evitar colisiones estéricas, las treonina proteasas hacen uso de su amida N-terminal como base, para aumentar la reactividad de la treonina catalítica.[19][20]
Ácida
El residuo acídico alinea y polariza al residuo básico. Por lo general este residuo ácido es un aspartato o glutamato. Algunas enzimas aún son activas poseyendo tan solo una díada, ya que el miembro ácido puede ser el menos necesario en las cisteína proteasas. Por ejemplo la papaína emplea a una asparragina como tercer miembro de la tríada, la cual orienta a la histidina, pero no actúa como ácido. En forma similar la proteasa de hepatitis A contiene una molécula de agua en la posición en la que debería estar el residuo ácido. Finalmente, las proteasas de citomegalovirus utilizan un par de histidinas, una actuando como la base (como es lo usual), y otra como el ácido.[1] La segunda histidina no es tan efectiva como ácido como lo son el glutamato o el aspartato, por lo que tiene una menor eficiencia catalítica.
Ejemplos de tríadas

Ser-His-Asp
La quimotripsina (superfamilia PA, Familia S1) se considera una de las enzimas que contiene una tríada clásica. Hace uso de un motivo serina-histidina-aspartato para producir la proteólisis. La quimotripsina se une a su sustrato, un bucle expuesto que contiene un residuo hidrofóbico de gran tamaño. El aspartato se encuentra unido a histidina por medio de un enlace de hidrógeno; lo que aumenta el pKa de su nitrógeno imidazólico desde 7 hasta aproximadamente 12. Esto le permite a la histidina el funcionar como una base general fuerte, y desprotonar a la serina. La serina a su vez, actúa como nucleófilo, atacando al carbono carbonílico y forzando al oxígeno carbonílico a aceptar un electrón, formando un intermediario tetraédrico. Este intermediario se estabiliza por un hueco de oxoanion, involucrando al esqueleto amida de la serina.
El colapso de este intermediario para regenerar el carbonilo, provoca que la histidina done su protón al nitrógeno unido al carbono alfa. El nitrógeno y el fragmento de péptido C-terminal unido dejan el sitio por difusión. Luego una molécula de agua dona un protón a la histidina y el grupo OH−
remanente atacan al carbono carbonílico, formando otro intermediario tetraédrico. El OH es un grupo saliente más pobre que el fragmento C-terminal, de modo que, cuando el intermediario tetraédrico colapsa nuevamente, el grupo serina se desprende, y gana nuevamente un protón desde la histidina. El grupo N-terminal del péptido escindido ahora abandona el sitio activo por difusión.
La misma tríada ha evolucionado también en las α/β hidrolasas tales como algunas lipasas y esterasas, sin embargo la quiralidad se encuentra invertida. Adicionalmente se ha encontrado que la acetil hidrolasa cerebral (la cual posee el mismo plegamiento que una proteína G pequeña) también posee esta tríada. La tríada equivalente serina-histidina-glutamato es la que utiliza la enzima acetilcolinesterasa.
Cys-His-Asp
Varias familias de cisteína proteasas hacen uso de esta tríada, por ejemplo la TEV proteasa (Superfamilia PA, Familia C4) y la papaína (Superfamilia CA, Familia C1). Esta tríada actúa en forma similar a las tríadas de las serina proteasas, con algunas notables diferencias que se discuten un poco más adelante. Todavía resulta poco claro cuan importante es la Asp de la tríada de la papaína para el mecanismo de catálisis, y varias cisteínas proteasas de hecho contienen díadas catalíticas (por ejemplo la proteasa del virus de la hepatitis A).
Ser-His-His
La tríada de la proteasa de citomegalovirus (Superfamilia SH, Familia S21) utiliza histidina cumpliendo tanto las funciones de ácido como de base. La remoción de la histidina ácida sólo resulta en una disminución de unas 10 veces en la actividad catalítica (comparada con la más de 10 000 veces en que se reduce la actividad de la quimiotripsina cuando se remueve el aspartato). Esta tríada ha sido interpretada como una posible forma de generar una enzima menos activa que sea capaz de controlar la tasa de escisión.[16]
Ser-Glu-Asp
Una tríada inusual que se encuentra en las seldolisina proteasas (Superfamilia SB, Familia S53). El bajo pKa del grupo carboxilato del glutamato implica que éste sólo puede actuar como la base de la tríada a pH muy bajos. Se ha hipotetizado que esta tríada es una adaptación a ambientes muy específicos tales como manantiales calientes (p. ej. kumamolisina) o a los lisosomas celulares (p. ej. tripeptidil peptidasa).[16]
Thr-Nterm
Las treonina proteasas, como las que se encuentran en la subunidad proteasa de los proteosomas (Superfamilia PB, Familia T1) y ornitina aciltransferasas (Superfamilia PE, Familia T5) hacen uso del grupo alcohol secundario de una treonina en una forma análoga al alcohol primario de la serina.[19][20] Sin embargo debido a la interferencia estérica del grupo metilo extra de la treonina, el miembro que funge como base de la tríada es el grupo amida N-terminal el cual polariza una molécula de agua, la cual a su vez, desprotona al alcohol catalítico para aumentar su reactividad.[1][16]
Ser-Nterm y Cys-Nterm
En una forma similar a las treonina proteasas, existen también configuraciones equivalentes "solo serina" y "solo cisteína", tales como las de la penicilina acilasa G (Superfamilia PB, Familia S45) y de la penicilina acilasa V (Superfamilia PB, Familia S59) las cuales se encuentran evolutivamente emparentadas con las proteasas de proteasoma. De la misma forma estas enzimas hacen uso del grupo amida N-terminal como base.[16]
Ser-cisSer-Lys
Esta inusual tríada aparece sólo en una superfamilia de las amidasas. En este caso, la lisina actúa polarizando a la serina del medio. La serina del medio entonces forma dos fuertes enlaces hidrógeno con la serina nucleofílica para activarla (una con el alcohol de la cadena lateral, y laotra con la amida del esqueleto). La serina del medio se mantiene en una orientación cis muy inusual para facilitar el preciso contacto con los otros dos residuos de la tríada. Esta tríada es además inusual en el hecho de que la lisina y la serina cis actúan ambas como base para activar a la serina catalítica, pero es la misma lisina la que también actúa como el miembro acídico y formando los contactos estructurales clave.[21]
Comparación de los mecanismos de las cisteína y serina hidrolasas

Esta sección referencia a las investigaciones hechas sobre las proteasas, sin embargo los mismos mecanismos y argumentos se aplicans a las serina y cisteína hidrolasas en general.
Las enzimas nucleofílicas hacen uso de un grupo interconectado de sitios activos para lograr la catálisis. La sofisticación de la red del sitio activo provoca que los residuos involucrados en la catálisis, y los residuos que se encuentran en conctacto con estos últimos, sean los más conservados evolutivamente dentro de sus familias de proteínas.[22] En las tríadas catalíticas, los nucleófilos más comunes son la serina (un alcohol) o cisteína (un tiol). Comparado con el oxígeno, el orbital d adicional del azufre le otorga mayor tamaño (0.4 Å más grande),[23] más blando, forma enlaces de mayor longitud (dC-X y dX-H 1.3 veces mayores) y posee un menor pKa (por 5 unidades).[24] Aunque en este punto se han resaltado las diferencias químicas entre las cisteína y serina proteasas en la química catalítica, similares efectos se presentan en las hidrolasas y transferasas en general.
El pKa de la cisteína es lo suficientemente bajo como para que algunas cisteína proteasas (por ejemplo la papaína) hayan mostrado poseer un ion tiolato S−
en la enzima en su estado basal[25] (a) y muchas incluso carecen del miembro acídico de la tríada (b). La serina es también más dependiente de otros residuos para reducir su pKa[24] para la desprotonación concertada con catálisis (c) para optimizar la orientación de los miembros ácido-base de las tríadas (d).[2] El bajo pKa de la cisteína funciona en contra de la resolución del primer intermediario tetraédrico ya que la reacción inversa e improductiva del ataque nucleofílico original es el producto de ruptura más favorable.[2] La base de la tríada, por lo tanto se encuentra preferentemente orientada para protonar al grupo saliente amida (e) para asegurar que es eliminado dejando al azufre unido covalentemente al extremo N-terminal del sustrato. Finalmente, la resolución del complejo acil-enzima (para liberar el extremo C-terminal del sustrato) requiere que la serina sea reprotonada (f) mientras que la cisteína puede salir como S−
.
Estéricamente, el azufre de la cisteína además posee enlaces más largos y un radio de Van der Waals de mayor tamaño como para encajar en el sitio activo[26] y un nucleófilo mutado puede terminar atrapado en orientaciones improductivas. Por ejemplo la estructura cristalina de la tio-tripsina indica que la cisteína apunta alejándose del sustrato, en lugar de formar interacciones con el hueco de oxoanion.[23]
La especialización evolutiva de las enzimas en torno a las necesidades de su nucleófilo hace que no sea sorprendente que los nucleófilos no se puedan interconvertir en las proteasas existentes[25][27][28][29][30][31][32] (ni en la mayoría de las otras enzimas[33][34][35][36][37][38]) y las grandes reducciones en la actividad observadas (>104) no pueden ser explicadas como el resultado de una reactividad comprometida o un desalineamiento estructural.
Evolución divergente

A pesar de las diferencias químicas descritas un poco más arriba, es claro que algunas superfamilias de proteasas han evolucionado en forma divergente para usar diferentes nucleófilos. Esto puede ser inferido porque varias superfamilias (con plegamientos similares) contienen familias que hacen uso de diferentes nucleófilos, indicando que el cambio de nucleófilos ha ocurrido varias vecs durante la historia evolutiva, aunque el mecanismo evolutivo por el cual esto ocurre, todavía permanece poco claro.
Dentro de cada superfamilia de proteasas que contiene una mezcla de nucleófilos, (p. ej. el Clan PA), las familias se designan por su nucleófilo catalítico (C=cisteína proteasas, S=serina proteasas).
| Superfamilia | Familias | Ejemplos |
|---|---|---|
| Clan PA | C3, C4, C24, C30, C37, C62, C74, C99 | Proteasa TEV (Virus del grabado del tabaco) |
| S1, S3, S6, S7, S29, S30, S31, S32, S39, S46, S55, S64, S65, S75 | Quimotripsina (mamíferos, p.ej. bos taurus) | |
| Clan PB | C44, C45, C59, C69, C89, C95 | Precursor de la amidofosforribosiltransferasa (Homo sapiens) |
| S45, S63 | Precursor de la penicilina G acyiasa (Escherichia coli) | |
| T1, T2, T3, T6 | Componente del proteasoma de las archaeas, (Thermoplasma acidophilum) | |
| Clan PC | C26, C56 | Gama-glutamil hidrolasa (Rattus norvegicus) |
| S51 | Dipeptidasa E (Escherichia coli) | |
| Clan PD | C46 | Proteína hedgehog (Drosophila melanogaster) |
| N9, N10, N11 | Protón ATPasa tipo-V que contiene inteína subunidad catalítica A (Saccharomyces cerevisiae) | |
| Clan PE | P1 | DmpA aminopeptidasa (Ochrobactrum anthropi) |
| T5 | Precursor de la Ornitina acetiltransferasa (Saccharomyces cerevisiae) |
Evolución convergente

La enzimología de las proteasas provee algunos de los más claros ejemplos de evolución convergente. El mismo arreglo geométrico de los residuos de las tríadas han evolucionado independientemente más de 20 veces (en superfamilias de enzimas separadas) eso es porque existe un número limitado de arreglos productivos de los tres residuos de las tríadas, el esqueleto de la enzima, y el sustrato. Estos ejemplos reflejan las limitaciones intrínsecas en la química de las enzimas, impulsando a la evolución a converger independientemente en las mismas soluciones en forma repetida.[1][2]
Cisteína y serina hidrolasas
Las serina y cisteína proteasas hacen uso de diferentes grupos funcionales de aminoácidos (alcohol o tiol) como nucleófilo. Para activar a ese nucleófilo, estas enzimas orientan un residuo básico y uno ácido formando una tríada catalítica. Las limitaciones físicas y químicas de la catálisis enzimática han causado que los mismos arreglos en forma de tríada hayan evolucionado independientemente más de veinte veces en diferentes superfamilias de enzimas.[2]
La misma geometría de las tríadas han convergido en las serina proteasas tales como en las familias de la quimiotripsina y subtilisina. En forma similar, lo mismo ha ocurrido con las cisteína proteasas tales como en las superfamilias de la proteasa C3 viral y la papaína. Debido a las similitudes mecanísticas entre las serina y cisteína proteasas, todas estas tríadas han convergido hacia casi el mismo arreglo.
| Superfamilia | Familia de proteínas de las cisteína proteasas | Ejemplos |
|---|---|---|
| CA | C1, C2, C6, C10, C12, C16, C19, C28, C31, C32, C33, C39, C47, C51, C54, C58, C64, C65, C66, C67, C70, C71, C76, C78, C83, C85, C86, C87, C93, C96, C98, C101 | Papaína (Carica papaya) y calpaína (Homo sapiens) |
| CD | C11, C13, C14, C25, C50, C80, C84 | Caspasa-1 (Rattus norvegicus) y separasa (Saccharomyces cerevisiae) |
| CE | C5, C48, C55, C57, C63, C79 | Adenaína (adenovirus humano tipo 2) |
| CF | C15 | Piroglutamil-peptidasa I (Bacillus amyloliquefaciens) |
| CL | C60, C82 | Sortasa A (Staphylococcus aureus) |
| CM | C18 | Peptidasa 2 del virus de la hepatitis C (Virus de la hepatitis C) |
| CN | C9 | Proteasa tipo-nsP2 del virus sindbis (sindbis virus) |
| CO | C40 | dipeptidil-peptidasa VI (Lysinibacillus sphaericus) |
| CP | C97 | DeSI-1 peptidasa (Mus musculus) |
| PA | C3, C4, C24, C30, C37, C62, C74, C99 | Proteasa TEV (Virus del grabado del tabaco) |
| PB | C44, C45, C59, C69, C89, C95 | Precursor de la amidofosforribosiltransferasa (Homo sapiens) |
| PC | C26, C56 | gama-glutamil hidrolasa (Rattus norvegicus) |
| PD | C46 | Proteína hedgehog (Drosophila melanogaster) |
| PE | P1 | Aminopeptidasa DmpA (Ochrobactrum anthropi) |
| Sin asignar | C7, C8, C21, C23, C27, C36, C42, C53, C75 |
| Superfamilia | familia de proteínas de las serina proteasa | Ejemplos |
|---|---|---|
| PA | S1, S3, S6, S7, S29, S30, S31, S32, S39, S46, S55, S64, S65, S75 | quimotripsina A (Bos taurus) |
| PB | S45, S63 | Precursor de la penicilina G acilasa (Escherichia coli) |
| PC | S51 | dipeptidasa E (Escherichia coli) |
| PE | P1 | DmpA aminopeptidasa (Ochrobactrum anthropi) |
| SB | S8, S53 | subtilisina (Bacillus licheniformis) |
| SC | S9, S10, S15, S28, S33, S37 | prolil oligopeptidasa (Sus scrofa) |
| SE | S11, S12, S13 | D-Ala-D-Ala peptidasa C (Escherichia coli) |
| SF | S24, S26 | Peptidasa señal I (Escherichia coli) |
| SH | S21, S73, S77, S78, S80 | Ensamblina de citomegalovirus (herpesvirus 5 humano) |
| SJ | S16, S50, S69 | Peptidasa Lon-A (Escherichia coli) |
| SK | S14, S41, S49 | Peptidasa Clp (Escherichia coli) |
| SO | S74 | Endosialidasa CIMCD de fago K1F proteína autohidrolítica (fago K1F de Enterobacteria) |
| SP | S59 | Nucleoporina 145 (Homo sapiens) |
| SR | S60 | Lactoferrina (Homo sapiens) |
| SS | S66 | Mureína tetrapeptidasa LD-carboxipeptidasa (Pseudomonas aeruginosa) |
| ST | S54 | romboide-1 (Drosophila melanogaster) |
| Sin asignar | S48, S62, S68, S71, S72, S79, S81 |
Treonina proteasa

Las treonina proteasas hacen uso del aminoácido treonina como nucleófilo catalítico. A diferencia de la cisteína y serina, la treonina es un alcohol secundario (posee un grupo metilo). El grupo metilo de la treonina provoca una gran restricción a las posibles orientaciones de la tríada y el sustrato ya que el grupo metilo choca ya sea con el esqueleto de la enzima o con la base histidina. En consecuencia, la mayor parte de las treonina proteasas hacen uso de una treonina N-terminal para evitar los efectos de choque estérico.
Varias superfamilias de enzimas evolutivamente independientes con diferentes plegamientos proteicos, hacen uso del residuo treonina N-terminal como nucleófilo. Primeramente aparece en la Superfamilia PB (proteasomas que hacen uso del plegamiento Ntn)[19] y en segundo lugar en la Superfamilia PE (acetiltransferasas que utilizan el plegamiento DOM)[20] Esta concordancia en los sitios activos en proteínas con plegamientos completamente diferentes indican que el sitio activo ha evolucionado en forma convergente en diferentes superfamilias.[2][16]
| Superfamilia | Familia de proteínas de las treonina proteasas | Ejemplos |
|---|---|---|
| Clan PB | T1, T2, T3, T6 | proteasoma de archaea, componente beta (Thermoplasma acidophilum) |
| Clan PE | T5 | ornitine acetiltransferasa (Saccharomyces cerevisiae) |
Véase también
- Catálisis enzimática
- Proteólisis
- Proteasa
- Grupos funcionales
- Superfamilia de enzimas
- Clan PA
- Evolución convergente
- Evolución divergente
Referencias
- Dodson, G; Wlodawer, A (septiembre de 1998). «Catalytic triads and their relatives.». Trends in Biochemical Sciences 23 (9): 347-52. PMID 9787641. doi:10.1016/S0968-0004(98)01254-7.
- Buller, AR; Townsend, CA (19 de febrero de 2013). «Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad». Proceedings of the National Academy of Sciences of the United States of America 110 (8): E653-61. Bibcode:2013PNAS..110E.653B. PMID 23382230. doi:10.1073/pnas.1221050110.
- Perutz, Max (1992). Protein structure. New approaches to disease and therapy. Nueva York: W.H. Freeman and Co.
- Neurath, H (Oct 1994). «Proteolytic enzymes past and present: the second golden era. Recollections, special section in honor of Max Perutz». Protein Sci 3 (10): 1734-9. PMC 2142620. PMID 7849591. doi:10.1002/pro.5560031013.
- Ohman, KP; Hoffman, A; Keiser, HR (April 1990). «Endothelin-induced vasoconstriction and release of atrial natriuretic peptides in the rat.». Acta physiologica Scandinavica 138 (4): 549-56. PMID 2141214. doi:10.1111/j.1748-1716.1990.tb08883.x.
- Dixon, Gordon H.; Kauffman, Dorothy L.; Neurath, Hans (5 de marzo de 1958). «Amino Acid Sequence in the Region of Diisopropyl Phosphoryl Binding in Dip-Trypsin». Journal of the American Chemical Society 80 (5): 1260-1261. doi:10.1021/ja01538a059.
- WALSH, KA; NEURATH, H (October 1964). «TRYPSINOGEN AND CHYMOTRYPSINOGEN AS HOMOLOGOUS PROTEINS». Proceedings of the National Academy of Sciences of the United States of America 52 (4): 884-9. Bibcode:1964PNAS...52..884W. PMC 300366. PMID 14224394. doi:10.1073/pnas.52.4.884.
- de Haën, C; Neurath, H; Teller, DC (25 de febrero de 1975). «The phylogeny of trypsin-related serine proteases and their zymogens. New methods for the investigation of distant evolutionary relationships.». Journal of Molecular Biology 92 (2): 225-59. PMID 1142424. doi:10.1016/0022-2836(75)90225-9.
- Lesk, AM; Fordham, WD (10 de mayo de 1996). «Conservation and variability in the structures of serine proteinases of the chymotrypsin family.». Journal of Molecular Biology 258 (3): 501-37. PMID 8642605. doi:10.1006/jmbi.1996.0264.
- Blow, DM; Birktoft, JJ; Hartley, BS (Jan 25, 1969). «Role of a buried acid group in the mechanism of action of chymotrypsin». Nature 221 (5178): 337-40. Bibcode:1969Natur.221..337B. PMID 5764436. doi:10.1038/221337a0.
- Gorbalenya, AE; Blinov, VM; Donchenko, AP (Jan 6, 1986). «Poliovirus-encoded proteinase 3C: a possible evolutionary link between cellular serine and cysteine proteinase families.». FEBS Letters 194 (2): 253-7. PMID 3000829. doi:10.1016/0014-5793(86)80095-3.
- Bazan, JF; Fletterick, RJ (November 1988). «Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: structural and functional implications». Proceedings of the National Academy of Sciences of the United States of America 85 (21): 7872-6. Bibcode:1988PNAS...85.7872B. PMC 282299. PMID 3186696. doi:10.1073/pnas.85.21.7872.
- Phan, J; Zdanov, A; Evdokimov, AG; Tropea, JE; Peters HK, 3rd; Kapust, RB; Li, M; Wlodawer, A; Waugh, DS (Dec 27, 2002). «Structural basis for the substrate specificity of tobacco etch virus protease». The Journal of Biological Chemistry 277 (52): 50564-72. PMID 12377789. doi:10.1074/jbc.M207224200.
- Rawlings, N.D.; Barrett, A.J. (1993). «Evolutionary families of peptidases». Biochem J 290: 205-218.
- Rawlings ND, Barrett AJ, Bateman A; Barrett; Bateman (January 2010). «MEROPS: the peptidase database». Nucleic Acids Res. 38 (Database issue): D227-33. PMC 2808883. PMID 19892822. doi:10.1093/nar/gkp971.
- Ekici, OD; Paetzel, M; Dalbey, RE (December 2008). «Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration.». Protein science : a publication of the Protein Society 17 (12): 2023-37. PMID 18824507. doi:10.1110/ps.035436.108.
- Damblon, C; Raquet, X; Lian, LY; Lamotte-Brasseur, J; Fonze, E; Charlier, P; Roberts, GC; Frère, JM (5 de marzo de 1996). «The catalytic mechanism of beta-lactamases: NMR titration of an active-site lysine residue of the TEM-1 enzyme». Proceedings of the National Academy of Sciences of the United States of America 93 (5): 1747-52. Bibcode:1996PNAS...93.1747D. PMID 8700829. doi:10.1073/pnas.93.5.1747.
- Jelsch, C; Lenfant, F; Masson, JM; Samama, JP (9 de marzo de 1992). «Beta-lactamase TEM1 of E. coli. Crystal structure determination at 2.5 A resolution.». FEBS Letters 299 (2): 135-42. PMID 1544485. doi:10.1016/0014-5793(92)80232-6.
- Brannigan, JA; Dodson, G; Duggleby, HJ; Moody, PC; Smith, JL; Tomchick, DR; Murzin, AG (23 de noviembre de 1995). «A protein catalytic framework with an N-terminal nucleophile is capable of self-activation». Nature 378 (6555): 416-9. Bibcode:1995Natur.378..416B. PMID 7477383. doi:10.1038/378416a0.
- Cheng, H; Grishin, NV (July 2005). «DOM-fold: a structure with crossing loops found in DmpA, ornithine acetyltransferase, and molybdenum cofactor-binding domain.». Protein science : a publication of the Protein Society 14 (7): 1902-10. PMID 15937278. doi:10.1110/ps.051364905.
- Shin, S; Yun, YS; Koo, HM; Kim, YS; Choi, KY; Oh, BH (4 de julio de 2003). «Characterization of a novel Ser-cisSer-Lys catalytic triad in comparison with the classical Ser-His-Asp triad.». The Journal of Biological Chemistry 278 (27): 24937-43. PMID 12711609. doi:10.1074/jbc.M302156200.
- Halabi, N; Rivoire, O; Leibler, S; Ranganathan, R (Aug 21, 2009). «Protein sectors: evolutionary units of three-dimensional structure.». Cell 138 (4): 774-86. PMID 19703402. doi:10.1016/j.cell.2009.07.038.
- McGrath, ME; Wilke, ME; Higaki, JN; Craik, CS; Fletterick, RJ (28 de noviembre de 1989). «Crystal structures of two engineered thiol trypsins.». Biochemistry 28 (24): 9264-70. PMID 2611228. doi:10.1021/bi00450a005.
- Polgár, L; Asbóth, B (Aug 7, 1986). «The basic difference in catalyses by serine and cysteine proteinases resides in charge stabilization in the transition state.». Journal of Theoretical Biology 121 (3): 323-6. PMID 3540454. doi:10.1016/s0022-5193(86)80111-4.
- Beveridge, AJ (July 1996). «A theoretical study of the active sites of papain and S195C rat trypsin: implications for the low reactivity of mutant serine proteinases». Protein science : a publication of the Protein Society 5 (7): 1355-65. PMC 2143470. PMID 8819168. doi:10.1002/pro.5560050714.
- Abrahmsén, L; Tom, J; Burnier, J; Butcher, KA; Kossiakoff, A; Wells, JA (Apr 30, 1991). «Engineering subtilisin and its substrates for efficient ligation of peptide bonds in aqueous solution.». Biochemistry 30 (17): 4151-9. PMID 2021606. doi:10.1021/bi00231a007.
- Neet, KE; Koshland DE, Jr (November 1966). «The conversion of serine at the active site of subtilisin to cysteine: a "chemical mutation"». Proceedings of the National Academy of Sciences of the United States of America 56 (5): 1606-11. Bibcode:1966PNAS...56.1606N. PMID 5230319. doi:10.1073/pnas.56.5.1606.
- Turkenburg, Johan P.; Lamers, Marieke B. A. C.; Brzozowski, A. Marek; Wright, Lisa M.; Hubbard, Roderick E.; Sturt, Simone L.; Williams, David H. (21 de febrero de 2002). «Structure of a Cys25→Ser mutant of human cathepsin S». Acta Crystallographica Section D Biological Crystallography 58 (3): 451-455. doi:10.1107/S0907444901021825.
- Polgár, L; Asbóth, B (7 de agosto de 1986). «The basic difference in catalyses by serine and cysteine proteinases resides in charge stabilization in the transition state.». Journal of Theoretical Biology 121 (3): 323-6. PMID 3540454. doi:10.1016/s0022-5193(86)80111-4.
- Lawson, MA; Semler, BL (15 de noviembre de 1991). «Poliovirus thiol proteinase 3C can utilize a serine nucleophile within the putative catalytic triad». Proceedings of the National Academy of Sciences of the United States of America 88 (22): 9919-23. Bibcode:1991PNAS...88.9919L. PMID 1658804. doi:10.1073/pnas.88.22.9919.
- Cheah, KC; Leong, LE; Porter, AG (5 de mayo de 1990). «Site-directed mutagenesis suggests close functional relationship between a human rhinovirus 3C cysteine protease and cellular trypsin-like serine proteases.». The Journal of Biological Chemistry 265 (13): 7180-7. PMID 2158990.
- Hahn, C. S.; Strauss, J. H. (Jun 1990). «Site-directed mutagenesis of the proposed catalytic amino acids of the Sindbis virus capsid protein autoprotease». J Virol 64 (6): 3069-73. PMC 249494. PMID 2335827.
- Kowal, AT; Werth, MT; Manodori, A; Cecchini, G; Schröder, I; Gunsalus, RP; Johnson, MK (26 de septiembre de 1995). «Effect of cysteine to serine mutations on the properties of the [4Fe-4S] center in Escherichia coli fumarate reductase.». Biochemistry 34 (38): 12284-93. PMID 7547971. doi:10.1021/bi00038a024.
- Sigal, IS; Harwood, BG; Arentzen, R (December 1982). «Thiol-beta-lactamase: replacement of the active-site serine of RTEM beta-lactamase by a cysteine residue». Proceedings of the National Academy of Sciences of the United States of America 79 (23): 7157-60. Bibcode:1982PNAS...79.7157S. PMID 6818541. doi:10.1073/pnas.79.23.7157.
- Amara, AA; Rehm, BH (1 de septiembre de 2003). «Replacement of the catalytic nucleophile cysteine-296 by serine in class II polyhydroxyalkanoate synthase from Pseudomonas aeruginosa-mediated synthesis of a new polyester: identification of catalytic residues.». The Biochemical journal 374 (Pt 2): 413-21. PMID 12924980. doi:10.1042/BJ20030431.
- Walker, Ian; Easton, Christopher J.; Ollis, David L. (1 de enero de 2000). «Site-directed mutagenesis of dienelactone hydrolase produces dienelactone isomerase». Chemical Communications (8): 671-672. doi:10.1039/b000365o.
- Li, J; Szittner, R; Derewenda, ZS; Meighen, EA (Aug 6, 1996). «Conversion of serine-114 to cysteine-114 and the role of the active site nucleophile in acyl transfer by myristoyl-ACP thioesterase from Vibrio harveyi.». Biochemistry 35 (31): 9967-73. PMID 8756458. doi:10.1021/bi9605292.
- Sharp, JD; Pickard, RT; Chiou, XG; Manetta, JV; Kovacevic, S; Miller, JR; Varshavsky, AD; Roberts, EF; Strifler, BA; Brems, DN (16 de septiembre de 1994). «Serine 228 is essential for catalytic activities of 85-kDa cytosolic phospholipase A2.». The Journal of Biological Chemistry 269 (37): 23250-4. PMID 8083230.
- Lehninger, Principles of Biochemistry, 4th ed.
- Wilson, Eisner, Briggs, Dickerson, Metzenberg, O'Brien, Susman, Boggs, Life on Earth (c 1973, Sinauer Associates, Inc., Publisher, Stamford, Connecticut. ISBN 0-87893-934-2)
| Control de autoridades |
|
|---|
 Datos: Q751908
Datos: Q751908 Multimedia: Catalytic triads / Q751908
Multimedia: Catalytic triads / Q751908