Agrégation des protéines
L'agrégation des protéines (ou agrégation/agglomération protéique) est un phénomène biologique dans lequel les protéines mal repliées s'agrègent (c'est-à-dire qu'elles s'accumulent et se groupent entre elles), de façon intra- comme extra-cellulaire[1],[2]. Ces agrégats de protéines sont souvent corrélés avec des maladies. De fait, les agrégats de protéines ont été impliqués dans une grande variété de maladies qu'on a appelées amyloïdoses, parmi lesquelles on retrouve la SLA, les maladies d'Alzheimer et de Parkinson, ou encore les maladies à prions[3],[4].
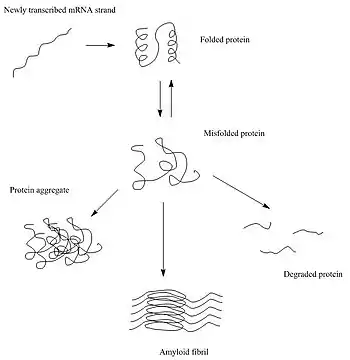
Après leur synthèse, les protéines ont pour habitude de se replier en une conformation tri-dimensionnelle particulière qui leur est le plus thermodynamiquement favorable : leur état natif[5]. Ce processus de repliement est entraîné par l'effet hydrophobe, qui est la tendance des parties hydrophobes de la protéine à essayer de se protéger de l'environnement hydrophile de la cellule en se terrant vers l'intérieur de la protéine. Ainsi, l'extérieur de la protéine est typiquement hydrophile, alors que son intérieur est typiquement hydrophobe.
Les structures protéiques sont stabilisées par des liaisons non covalentes et des ponts disulfure liant deux résidus cystéine. Les interactions non covalentes comprennent les liaisons ioniques et les interactions faibles de van der Waals. Les interactions ioniques se forment entre un anion et un cation et constituent des ponts salins aidant à stabiliser la protéine. Les interactions de van der Waals incluent les interactions non polaires (forces de London) et les interactions polaires (liaisons hydrogène, forces intermoléculaires). Celles-ci jouent un rôle important dans la structure secondaire des protéines, comme en formant une hélice alpha ou un feuillet bêta, ainsi que dans leur structure tertiaire. Les interactions entre les résidus d'acides aminés dans une protéine donnée sont très importantes pour la structure finale de cette protéine.
Lorsqu'il y a des changements au sein des interactions non covalentes, comme il peut en survenir à la faveur d'un changement dans la séquence en acides aminés, la protéine a des chances de mal se replier ou ne pas se replier du tout. Dans ce type de situation, si la cellule n'aide pas les protéines à corriger ce défaut de repliement ou ne les détruit pas, elles peuvent s'agglomérer, et dans ce processus les parties hydrophobes exposées des protéines peuvent interagir avec les parties hydrophobes exposées d'autres protéines non défectueuses[6],[7]. Il existe trois grands types d'agrégats protéiques qui peuvent se former : les agrégats amorphes, les oligomères et les fibrilles amyloïdes[8].
Causes
L'aggrégation des protéines peut avoir lieu pour plusieurs causes. Il existe quatre classes dans lesquelles ces causes peuvent être catégorisées, qui sont détaillées ci-dessous.
Mutations
Les mutations se produisant dans la séquence d'ADN peuvent affecter ou non la séquence en acides aminés de la protéine considérée. Si la séquence est affectée, un acide aminé différent peut changer les interactions entre les chaînes latérales, ce qui a des conséquences sur le repliement de la protéine. Cela peut conduire à une exposition des régions hydrophobes de la protéine qui s'agrègent avec la même protéine mal repliée ou non repliée, ou bien avec une protéine différente.
En plus des mutations affectant les protéines elles-mêmes, l'agrégation protéique peut aussi être causée indirectement par des mutations survenant dans les protéines au cours de voies de régulation comme la voie de repliement (chaperones) ou la voie ubiquitine-protéasome (ubiquitine ligases)[9]. Les chaperones aident les protéines à se replier en leur fournissant un environnement sûr pour se replier. Les ubiquitine ligases ciblent les protéines à dégrader par une modification de l'ubiquitine.
Défauts au cours de la synthèse protéique
L'agrégation protéique peut être causée par des problèmes survenant lors de la transcription ou de la traduction. Lors de la transcription, l'ADN est copié en ARNm, formant un brin de pré-ARNm qui subit des modifications post-transcriptionnelles pour former un ARNm[10]. Lors de la traduction, les ribosomes et les ARnt permettent de traduire la séquence d'ARNm en séquence d'acides aminés[10]. Si des défauts surviennent au cours de l'une de ces deux étapes, produisant un brin d'ARNm ou une séquence en acides aminés incorrects, cela cause un mauvais repliement de la protéine, menant à une agrégation protéique.
Stress environnementaux
Les stress environnementaux comme des températures ou un pH extrêmes, ou bien un stress oxydatif peuvent également conduire à une agrégation protéique[11]. Un exemple de maladie causée par ce type de stress est la cryoglobulinémie.
Les températures extrêmes peuvent affaiblir et déstabiliser les liaisons non covalentes entre les résidus d'acides aminés. Les pH extérieurs à la gamme de pH d'une protéine donnée peuvent modifier l'état de protonation des acides aminés, ce qui peut augmenter ou diminuer les interactions non covalentes. Cela peut également conduire à des liaisons moins stables et par conséquent au non repliement de la protéine.
Le stress oxydatif peut être causé par des radicaux tels que les ROS. Ces radicaux instables peuvent attaquer les résidus d'acides aminés, ce qui conduit à l'oxydation des chaînes latérales (comme les chaînes aromatiques latérales ou les chaînes de méthionine latérales) et/ou au clivage des liaisons polypeptides[12]. Cela peut affecter les liaisons non covalentes qui permettent de tenir la protéine dans une forme correcte, ce qui peut causer une déstabilisation de la protéine, et ainsi empêcher la protéine de se replier[11].
Vieillissement
Les cellules possèdent des mécanismes qui peuvent replier une nouvelle fois ou bien dégrader les agrégats protéiques. Cependant, quand les cellules vieillissent, ces mécanismes de contrôle sont moins performants et la cellule est moins capable de prendre en charge ces agrégats[11].
L'hypothèse selon laquelle l'agrégation protéique est un processus conséquent au vieillissement est désormais testable car certains modèles de vieillissement retardé sont en cours d'élaboration. Si le développement d'agrégats protéiques était un processus indépendant du vieillissement, le fait de ralentir ce vieillissement n'aurait aucun effet sur le taux de protéotoxicité au cours du temps. Cependant, si le vieillissement était associé à un déclin dans l'activité des mécanismes de protection contre la protéotoxicité, les modèles de vieillissement ralenti montreraient une réduction dans l'agrégation et la protéotoxicité. Afin de résoudre ce problème, plusieurs tests de toxicité ont été réalisés chez le nématode C. elegans. Ces études ont montré que la réduction de l'activité du signalement insuline/IGF, une voie de régulation du vieillissement bien connue, protège de l'agrégation protéique toxique liée à la neurodégénération. La validité de cette approche a été testée et confirmée chez les mammifères, car le fait de réduire l'activité de la voie de signalisation IGF-1 a protégé des souris d'étude atteintes d'Alzheimer de présenter un comportement et des déficiences associées à cette maladie[13].
Localisation des agrégats
Plusieurs études ont montré que les réponses cellulaires à l'agrégation protéique sont bien régulées et organisées. Les agrégats protéiques sont localisés dans des zones spécifiques de la cellule, et la recherche s'est focalisée sur ces zones chez des procaryotes (E. coli) et des eucaryotes (levures, cellules mammaliennes).
Chez les bactéries
Les agrégats des bactéries se retrouvent de façon asymétrique à l'un des pôles de la cellule, le "pôle plus antérieur" (older pole en anglais). À la suite de la division cellulaire, la cellule fille contenant le pôle plus antérieur obtient l'agrégat protéique et croît de façon plus lente que la cellule fille sans l'agrégat. Cela constitue un mécanisme de sélection naturelle pour réduire les agrégats protéiques au sein de la population bactérienne[14].
Chez les levures
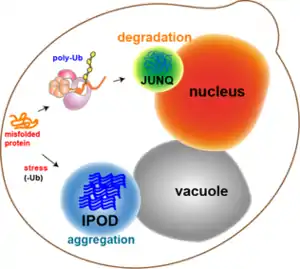
La plupart des agrégats protéiques chez les cellules de levure voient leur défaut de repliement corrigé par les chaperones. Cependant, certains agrégats peuvent ne pas voir leur défaut de repliement corrigé, comme les protéines endommagées par un stress oxydatif ou les protéines destinées à la dégradation. À la place, elles peuvent être redirigées vers l'un des deux compartiments suivants : le compartiment de contrôle qualité juxtanucléaire (JUxtaNuclear Quality-control compartment ou JUNQ en anglais) près de la membrane nucléaire, et le dépôt de protéines insolubles (Insoluble Protein Deposit ou IPOD en anglais) près de la vacuole dans les cellules de levure[11]. Les agrégats protéiques vont dans le JUNQ lorsqu'ils sont ubiquitinés et destinés à être dégradés, alors que les protéines agglomérées et insolubles vont dans l'IPOD pour un stockage plus long. Des résultats ont montré que les protéines à cet endroit peuvent en être retirées par autophagie[15]. Ces deux possibilités fonctionnent ensemble de telle façon que les protéines tendent à aller vers l'IPOD lorsque la voie du protéasome est surchargée[15].
Chez les cellules mammaliennes
Dans les cellules mammaliennes, ces agrégats protéiques sont appelés « agrésomes » et se forment lorsque la cellule est malade. Cela arrive car les agrégats tendent à se former lorsque des protéines hétérologues sont présentes dans la cellule, ce qui peut se produire lorsque la cellule est mutée. L'ubiquitine ligase E3 est capable de reconnaître des protéines mal repliées et de les ubiquitiner. La HDAC6 peut ensuite lier l'ubiquitine et la protéine motrice dynéine afin de transporter les agrégats « marqués » jusqu'au centrosome. Une fois là-bas, ils se groupent ensemble dans une sphère qui entoure le centrosome. Ils apportent les chaperones et les protéasomes et activent l'autophagie[16].
Élimination des agrégats
Il existe deux principaux systèmes de "contrôle qualité" dans la cellule responsables de l'élimination des agrégats protéiques. Les protéines mal repliées peuvent être repliées à nouveau par le système bi-chaperon ou dégradées par le système ubiquitine-protéasome, ou par autophagie[17].
Néo-repliement
Le système bi-chaperone utilise les chaperones Hsp70 (DnaK-DnaJ-GrpE chez E. coli et Ssa1-Ydj1/Sis1-Sse1/Fe1 chez la levure) et Hsp100 (ClpB chez E. coli et Hsp104 chez la levure) pour désagglomérer et néo-replier les protéines[18].
La Hsp70 interagit avec des agrégats protéiques et recrute la Hsp100. La Hsp70 stabilise une Hsp100 activée. Les Hsp100 possèdent des boucles de pores aromatiques utilisées pour l'activité de threading (fil conducteur) pour démêler les polypeptides simples. Cette activité de threading peut être initiée à l'extrémité N-terminale, C-terminale ou bien au milieu du polypeptide. Le polypeptide subit une translocation par la Hsp100 au cours d'une série d'étapes, chacune d'entre elles consommant un ATP. Le polypeptide ne se replie pas et se néo-replie ensuite soit par lui-même soit avec l'aide de protéines de choc thermique (heat shock proteins)[19].
Dégradation
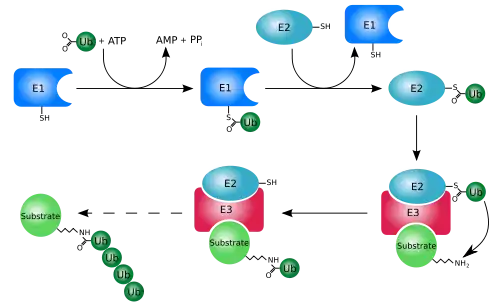
Les protéines mal repliées peuvent être éliminées par le système ubiquitine-protéasome. Cela consiste en une voie E1-E2-E3 qui ubiquitine les protéines pour les destiner à la dégradation. Chez les eucaryotes, les protéines sont dégradées par le protéasome 26S. Chez les cellules mammaliennes, la ligase E3 et la protéine carboxy-terminale interagissant avec la Hsp70 ciblent les protéines liées à la Hsp70. Chez la levure, les ligases E3 Doa10 et Hrd1 ont des fonctions similaires sur les protéines du réticulum endoplasmique[20] :
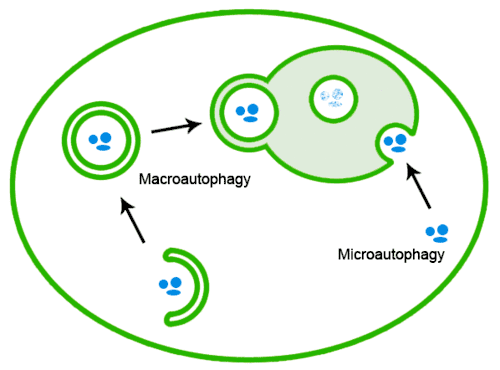
Les protéines mal repliées peuvent également être éliminées par l'autophagie, lors de laquelle les agrégats protéiques sont transportés jusqu'au lysosome[20] :
Toxicité
Bien qu'on pensait que les agrégats protéiques matures soient toxiques en tant que tels, des résultats récents suggèrent qu'en fait ce sont les agrégats protéiques immatures qui sont les plus toxiques[21],[22]. Les groupes hydrophobes de ces agrégats peuvent interagir avec d'autres composants de la cellule et les endommager. Les hypothèses sont que la toxicité des agrégats protéiques est liée aux mécanismes de séquestration des composants cellulaires, à la génération de ROS, à la liaison à des récepteurs spécifiques dans la membrane ou par la perturbation des membranes[23]. Un test génétique a permis de déterminer que les espèces ayant un poids moléculaire plus élevé sont responsables de la perméabilisation de la membrane[24]. Il est connu que les agrégats protéiques peuvent in vitro déstabiliser des bicouches lipidiques artificielles, ce qui conduit à la perméabilisation de la membrane.
Voir aussi
Notes
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Protein aggregation » (voir la liste des auteurs).
Références
- Adriano Aguzzi et Tracy O'Connor, « Protein aggregation diseases: pathogenicity and therapeutic perspectives », Nature Reviews. Drug Discovery, vol. 9, no 3, , p. 237–248 (ISSN 1474-1784, PMID 20190788, DOI 10.1038/nrd3050, lire en ligne, consulté le )
- Massimo Stefani et Christopher M. Dobson, « Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution », Journal of Molecular Medicine (Berlin, Germany), vol. 81, no 11, , p. 678–699 (ISSN 0946-2716, PMID 12942175, DOI 10.1007/s00109-003-0464-5, lire en ligne, consulté le )
- Fernanda G. De Felice, Marcelo N. N. Vieira, M. Nazareth L. Meirelles et Ludmilla A. Morozova-Roche, « Formation of amyloid aggregates from human lysozyme and its disease-associated variants using hydrostatic pressure », FASEB journal: official publication of the Federation of American Societies for Experimental Biology, vol. 18, no 10, , p. 1099–1101 (ISSN 1530-6860, PMID 15155566, DOI 10.1096/fj.03-1072fje, lire en ligne, consulté le )
- Rudolph E. Tanzi et Lars Bertram, « Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective », Cell, vol. 120, no 4, , p. 545–555 (ISSN 0092-8674, PMID 15734686, DOI 10.1016/j.cell.2005.02.008, lire en ligne, consulté le )
- Ansgar Brüning et Julia Jückstock, « Misfolded proteins: from little villains to little helpers in the fight against cancer », Frontiers in Oncology, vol. 5, , p. 47 (ISSN 2234-943X, PMID 25759792, PMCID PMC4338749, DOI 10.3389/fonc.2015.00047, lire en ligne, consulté le )
- M. J. Gething et J. Sambrook, « Protein folding in the cell », Nature, vol. 355, no 6355, , p. 33–45 (ISSN 0028-0836, PMID 1731198, DOI 10.1038/355033a0, lire en ligne, consulté le )
- Christopher J. Roberts, « Non-native protein aggregation kinetics », Biotechnology and Bioengineering, vol. 98, no 5, , p. 927–938 (ISSN 0006-3592, PMID 17705294, DOI 10.1002/bit.21627, lire en ligne, consulté le )
- (en) David L. Cox et Michael M. Nelson, Lehninger Principles of Biochemistry, New York, W. H. Freeman, (ISBN 978-1-4292-3414-6), p. 143
- Sarah J Shoesmith Berke et Henry L Paulson, « Protein aggregation and the ubiquitin proteasome pathway: gaining the UPPer hand on neurodegeneration », Current Opinion in Genetics & Development, vol. 13, no 3, , p. 253–261 (DOI 10.1016/S0959-437X(03)00053-4, lire en ligne, consulté le )
- (en) Robert F. Weaver, Molecular Biology, New York, McGraw-Hill, (ISBN 978-0-07-352532-7), p. 122-156 ; 523-600
- Jens Tyedmers, Axel Mogk et Bernd Bukau, « Cellular strategies for controlling protein aggregation », Nature Reviews. Molecular Cell Biology, vol. 11, no 11, , p. 777–788 (ISSN 1471-0080, PMID 20944667, DOI 10.1038/nrm2993, lire en ligne, consulté le )
- (en) E. R. Stadtman et R. L. Levine, « Free radical-mediated oxidation of free amino acids and amino acid residues in proteins », Amino Acids, vol. 25, nos 3-4, , p. 207–218 (ISSN 0939-4451 et 1438-2199, DOI 10.1007/s00726-003-0011-2, lire en ligne, consulté le )
- James F. Morley, Heather R. Brignull, Jill J. Weyers et Richard I. Morimoto, « The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans », Proceedings of the National Academy of Sciences of the United States of America, vol. 99, no 16, , p. 10417–10422 (ISSN 0027-8424, PMID 12122205, DOI 10.1073/pnas.152161099, lire en ligne, consulté le )
- Natalia G. Bednarska, Joost Schymkowitz, Frederic Rousseau et Johan Van Eldere, « Protein aggregation in bacteria: the thin boundary between functionality and toxicity », Microbiology (Reading, England), vol. 159, no Pt 9, , p. 1795–1806 (ISSN 1465-2080, PMID 23894132, DOI 10.1099/mic.0.069575-0, lire en ligne, consulté le )
- Mari Takalo, Antero Salminen, Hilkka Soininen et Mikko Hiltunen, « Protein aggregation and degradation mechanisms in neurodegenerative diseases », American Journal of Neurodegenerative Disease, vol. 2, no 1, , p. 1–14 (ISSN 2165-591X, PMID 23516262, PMCID PMC3601466, lire en ligne, consulté le )
- (en) Rafael Garcia-Mata, Ya-Sheng Gao et Elizabeth Sztul, « Hassles with Taking Out the Garbage: Aggravating Aggresomes », Traffic, vol. 3, no 6, , p. 388–396 (ISSN 1600-0854, DOI 10.1034/j.1600-0854.2002.30602.x, lire en ligne, consulté le )
- (en) Niels Gregersen, Lars Bolund et Peter Bross, « Protein misfolding, aggregation, and degradation in disease », Molecular Biotechnology, vol. 31, no 2, , p. 141–150 (ISSN 1073-6085 et 1559-0305, DOI 10.1385/MB:31:2:141, lire en ligne, consulté le )
- Axel Mogk, Eva Kummer et Bernd Bukau, « Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation », Frontiers in Molecular Biosciences, vol. 2, , p. 22 (ISSN 2296-889X, PMID 26042222, PMCID PMC4436881, DOI 10.3389/fmolb.2015.00022, lire en ligne, consulté le )
- Krzysztof Liberek, Agnieszka Lewandowska et Szymon Zietkiewicz, « Chaperones in control of protein disaggregation », The EMBO journal, vol. 27, no 2, , p. 328–335 (ISSN 1460-2075, PMID 18216875, PMCID PMC2234349, DOI 10.1038/sj.emboj.7601970, lire en ligne, consulté le )
- Bryan Chen, Marco Retzlaff, Thomas Roos et Judith Frydman, « Cellular strategies of protein quality control », Cold Spring Harbor Perspectives in Biology, vol. 3, no 8, , a004374 (ISSN 1943-0264, PMID 21746797, PMCID PMC3140689, DOI 10.1101/cshperspect.a004374, lire en ligne, consulté le )
- Y. J. Zhu, H. Lin et R. Lal, « Fresh and nonfibrillar amyloid beta protein(1-40) induces rapid cellular degeneration in aged human fibroblasts: evidence for AbetaP-channel-mediated cellular toxicity », FASEB journal: official publication of the Federation of American Societies for Experimental Biology, vol. 14, no 9, , p. 1244–1254 (ISSN 0892-6638, PMID 10834946, lire en ligne, consulté le )
- C. Nilsberth, A. Westlind-Danielsson, C. B. Eckman et M. M. Condron, « The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation », Nature Neuroscience, vol. 4, no 9, , p. 887–893 (ISSN 1097-6256, PMID 11528419, DOI 10.1038/nn0901-887, lire en ligne, consulté le )
- (en) Claudio Soto, « Unfolding the role of protein misfolding in neurodegenerative diseases », Nature Reviews Neuroscience, vol. 4, no 1, , p. 49–60 (ISSN 1471-003X, DOI 10.1038/nrn1007, lire en ligne, consulté le )
- Patrick Flagmeier, Suman De, David C. Wirthensohn et Steven F. Lee, « Ultrasensitive Measurement of Ca(2+) Influx into Lipid Vesicles Induced by Protein Aggregates », Angewandte Chemie (International Ed. in English), vol. 56, no 27, , p. 7750–7754 (ISSN 1521-3773, PMID 28474754, PMCID PMC5615231, DOI 10.1002/anie.201700966, lire en ligne, consulté le )
Liens externes
- (en) Folding @ Home Aide au décodage du repliement des protéines.
- (en) Rosetta @ Home Aide à la prédiction de structures et des complexes protéiques.
 Portail de la biochimie
Portail de la biochimie  Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire