Facteur VIII
Le facteur VIII ou facteur anti-hémophilique A est une protéine contenue dans le plasma (sang débarrassé de ses globules rouges et de ses globules blancs) à l'état de traces, jouant un rôle de cofacteur dans la cascade de la coagulation.

Structure
L'analyse de la molécule facteur VIII montre qu'elle est constituée de trois domaines distincts A, B, C. Le domaine A de 330 AA est fait de trois exemplaires A1, A2, A3. Le domaine B de 983 AA est unique. Le domaine C fait de deux segments C1, C2 de 150 AA chacun. La séquence des différents domaines à partir de l'extrémité amino-terminale est A1-A2-B-A3-C1-C2. Il existe des homologies de structure entre : les domaines A du facteur VIII et celle du facteur V (30 à 40 %) et la céruloplasmine[1].
Gène du facteur VIII

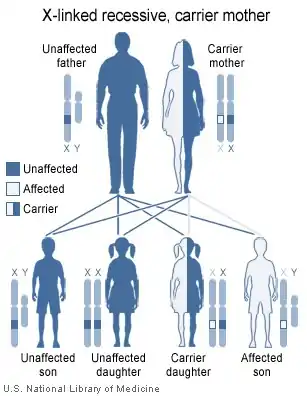
Le gène responsable de la fabrication du facteur VIII qui est défectueux (mutant) dans l'hémophilie, se situe sur l'extrémité distale du bras long du chromosome X. Cela fait de l'hémophilie une maladie héréditaire liée au sexe[2]. Il a une longueur de 186 000 paires de bases (bp), ce qui représente 0,1 % du chromosome X. Le gène codant l'ARNm de la protéine est constitué de 26 exons contenant 69 à 3 106 bp. Les 25 introns se répartissent entre 207 à 32 400 bp.
Synthèse
La source principale du facteur VIII semble être le foie. L'utilisation des anticorps monoclonaux a prouvé le rôle essentiel de l'hépatocyte dans cette synthèse. Par contre, l'augmentation du taux de facteur VIII dans les insuffisances hépato-cellulaires, suppose d'autres sources. En effet, ceci a pu être confirmé par la mise en évidence de l'ARNm de ce facteur au niveau d'autres organes, en particulier, le rein, la rate et au niveau des lymphocytes.
Production
La production du facteur VIII commence par la synthèse d'un polypeptide de 2 351 acides aminés. Au niveau du réticulum endoplasmique, la protéine perd son peptide signal de 19 AA et subit une série de glycosylation. La molécule finale a un poids moléculaire de 265 à 330 kDa, et contient 2 332 AA. Les protéines de transport (binding protein) vont se détacher de la molécule. Celle-ci, toujours monomérique, transite dans l'appareil de golgi où s'effectuent les modifications post-traductionnelle. Le monomère de 330 kDa va subir une coupure en un dimère, formé d'une chaîne lourde de 210 kDa et d'une chaîne légère de 80. Dans le plasma, le facteur VIII, stabilisé par le facteur de von Willebrand existe à un taux de 0,1 µg/ml. À l'état libre, il est rapidement clivé par les serines protéases, ce qui rend difficile son isolement et sa purification[3].
Fonction
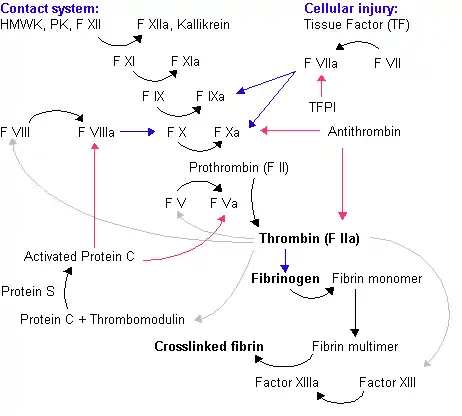
Le facteur VIII joue un rôle central dans la coagulation. Il s'agit du cofacteur de facteur IX. Le facteur VIII, activé par la thrombine, devient le catalyseur de la réaction d'activation du facteur X par le facteur IX activé, en présence d'ion calcium et de phospholipides. La réaction d'activation du facteur X est accélérée environ 200 000 fois en présence du facteur VIII. Le facteur X activé acquiert une activité catalytique qui lui permet de transformer la prothrombine en thrombine. Celle-ci dégrade le fibrinogène en fibrine. Le caillot ainsi formé sera stabilisé par le facteur XIII, ce qui permet l'arrêt du saignement[4].
Pathologie associée aux déficiences en facteur VIII
Le mot hémophilie vient de deux mots grecs : haïma, qui signifie sang et philia qui signifie affection. L'hémophilie A est une maladie héréditaire. Cela signifie que l'enfant naît avec la maladie. Le sang d'une personne atteinte d'hémophilie ne coagule pas normalement. Les saignements ne sont pas plus abondants, ni plus rapides que la normale, mais ils durent plus longtemps.
L'hémophilie A affecte moins d'une personne sur 10 000.
Symptômes de l'hémophilie A
- Hémorragie externe : l'hémorragie externe est visible puisque le sang s'écoule par une plaie localisée à la surface du corps. La personne peut saigner du nez à l'occasion d'un rhume ou après une exposition prolongée au soleil. Elle peut aussi saigner de la bouche : brossage des dents trop intense, morsure de la muqueuse des joues à l'occasion de la mastication…
- Hématome : l'hématome peut survenir à n'importe quel endroit du corps. Il est consécutif à un traumatisme s'accompagnant d'apparition de douleurs et d'un gonflement perceptible à la palpation, à l'endroit du traumatisme. Sa caractéristique principale est la survenue d'une coloration tout d'abord bleue violette qui se transforme progressivement en coloration jaune verdâtre qui disparaît au bout de 15 jours environ.
- Hémarthrose : le sang s'écoule à l'intérieur d'une articulation (genou, cheville, coude, doigt) et finit, en l'absence de traitement substitutif, par la « bloquer ». Cette hémorragie peut être provoquée par un traumatisme (coup, chute) ou faire suite à un effort prolongé (longue marche, pratique d'un sport, port d'une charge importante). Elle peut aussi être spontanée.
Sévérité
L’hémophilie A peut se diviser en trois classes :
- Les formes sévères sont le plus souvent diagnostiquées dès la naissance devant un hématome survenu spontanément ou après un traumatisme minime.
- Les formes modérées sont moins fréquentes et le plus souvent causées par un traumatisme même léger.
- Dans les formes légères, les hémorragies surviennent uniquement à la suite d’une blessure grave ou à un acte opératoire. Une intervention chirurgicale mineure (extraction dentaire) peut être à l’origine d’accidents hémorragiques.
Facteur VIII en tant que médication
Traitement de l'hémophilie
Le traitement de l'hémophilie A consiste en l'administration de facteurs de coagulation. Les facteurs de coagulation doivent être délivrés dans le sang par injection intraveineuse. Dans le cas d'hémorragie, le traitement par facteur VIII doit idéalement être administré le plus rapidement possible. Le processus de coagulation sanguine, ainsi corrigé, peut se faire normalement et l'hémorragie s'interrompre.
Le facteur VIII a été associés avec le facteur de Willebrand pour prolonger la durée l'action thérapeutique [5]. BIVV001 est une protéine obtenue par génie génétique consistant en une fusion du facteur VIII avec une immunoglobuline G1-Fc. Et un polypeptide 2-XTEN (Amunix Pharmaceuticals Inc). Cette nouvelle molécule est administrée par voie sous-cutanée et permet de prolonger la demi-vie du facteur VIII d'un facteur de trois à quatre [6]. Les études concernant ce nouveau médicament sont financées par Sanofi [7] .
Complications des traitements
Les complications majeurs des traitements sont :
- Les inhibiteurs : il arrive que l'organisme réagisse contre le facteur injecté en fabriquant des anticorps. On parle alors de la survenue d'un inhibiteur[8]. Un examen biologique permet de doser son taux.
- Allergie : les premiers concentrés de facteur VIII étaient fabriqués à partir de plasma sanguin et d'albumine d'origine humaine au cours de don du sang ou de plasma, potentiellement allergisant. Les traitements à la chaleur et l'élaboration de composants exempts de protéines d’origine humaine ou animale[9] ont permis de diminuer les risques de complication des traitements.
- La transmission d'agents infectieux : trois virus ont en effet eu des conséquences dramatiques chez les hémophiles : le virus de l'immunodéficience humaine (VIH), le virus de l'hépatite B et le virus de l'hépatite C (voir affaire du sang contaminé en France ainsi que dans d'autres pays. Les contaminations par ces virus ne sont plus observées en raison de la nature des procédés de fabrication récents, le facteur VIII étant produit actuellement par génie génétique et ne comprenant donc plus aucun élément issu de don du sang.
Enfin, le risque de contamination par de nouveaux agents infectieux comme les prions reste hypothétique en l'état actuel des connaissances.
Divers
Le facteur VIII fait partie de la liste des médicaments essentiels de l'Organisation mondiale de la santé (liste mise à jour en )[10].
Notes et références
- Fay PJ, Haidaris PJ, Smudzin TM. Human factor VIIIa subunit structure. Reconstruction of factor VIIIa from the isolated A1/A3-C1-C2 dimer and A2 subunit. J Biol Chem. 1991 May 15;266(14):8957–8962.
- http://www.orpha.net/data/patho/Pub/fr/Hemophilie-FRfrPub646.pdf
- Hultin MB, Jesty J. The activation and inactivation of human factor VIII by thrombin: effect of inhibitors of thrombin. Blood. 1981 Mar;57(3):476–482
- Theodore S. Zimmerman, Zaverio M. Ruggeri Coagulation and Bleeding Disorders: The Role of Factor VIII and Von willebrand factor. 1989 1:72 (ISBN 0-8247-8013-2)
- David Lillicrap Improvements in factor concentrates Curr Opin Hematol. 2010 Sep;17(5):393-76. doi:10.1097/MOH.0b013e32833c06c
- BIVV001 Fusion Protein as Factor VIII Replacement Therapy for Hemophilia A N Engl J Med 2020; 383:1018-1027 DOI: 10.1056/NEJMoa2002699.
- Pier M. Mannucci Treatment of Hemophilia — More Amazing ProgressN Engl J Med 2020; 383:1068-1070 DOI: 10.1056/NEJMe2024545
- Développement des inhibiteurs et prise en charge chez les patients hémophiles traités par Facteur VIII ou IX d'origine plasmatique ou recombinante
- Baxter Canada BioScience
- WHO Model List of Essential Medicines, 18th list, avril 2013
Voir aussi
Articles connexes
Liens externes
- (en) Facteur VIII: entrée dans la banque de données des proteines (PDB) permet de visualiser la structure à l'écran
- (en) RefSeq: banque de données biologiques, permet de visualiser la structure en mode dynamique du facteur VIII
- (en) GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005 Hémophilie A
 Portail de la médecine
Portail de la médecine  Portail de l’hématologie
Portail de l’hématologie  Portail de la pharmacie
Portail de la pharmacie