Groupe protecteur
En chimie organique on appelle groupe protecteur (ou groupement protecteur) un groupe fonctionnel introduit dans la molécule à partir d'une fonction chimique pour masquer tout ou partie de sa réactivité[1]. L'introduction d'un groupe protecteur a pour but d'améliorer la sélectivité des réactions suivantes.
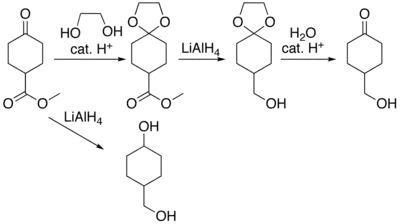
Lors d'une synthèse multi-étapes, il est courant de se retrouver aux prises avec des problèmes de chimiosélectivité lorsque plusieurs groupes fonctionnels peuvent réagir lors d'une même réaction. Dans ce cas, on essaye de transformer un ou plusieurs groupes fonctionnels en d'autres groupes qui seront inertes dans les conditions réactionnelles choisies. Cette étape s'appelle une protection. Une fois la réaction menée à bien en présence des groupes protecteurs, ces derniers sont à nouveau transformés pour revenir aux groupes fonctionnels de départ ; c'est la « déprotection ».
Principe

Un groupe fonctionnel doit respecter 7 critères[2] afin d'être considéré comme un bon groupe protecteur, empêchant ainsi sa destruction ou inhibant sa réactivité lors d'une réaction chimique :
- La méthode de protection doit être simple et efficace
- Le(s) réactif(s) utilisé(s) doi(ven)t être facilement accessible(s) (idéalement disponible(s) commercialement)
- Le groupe protecteur doit être facilement caractérisé, et éviter les complications lors de l'analyse (par exemple, en introduisant de nouveaux centres asymétriques)
- Il doit résister aux conditions usuelles de purification (colonne de chromatographie)
- Il doit être résistant à une large gamme de réactions chimiques
- Il doit pouvoir être retiré de manière douce et sélective, en employant des conditions très spécifiques
- Les sous-produits de la réaction de déprotection doivent pouvoir être facilement éliminés lors du processus de purification.
Déprotection
Orthogonalité
Lors d'une synthèse organique, plusieurs groupes protecteurs peuvent être présent simultanément sur un substrat. La stratégie de synthèse choisie va nécessiter la déprotection sélective et successive des différentes fonctions. Il importe donc de pouvoir éliminer un groupe protecteur sans modifier les autres groupes présents sur le substrat. Les différents groupes protecteurs sont classés dans des « ensembles orthogonaux »[N 1], qui sont des ensembles idéaux regroupant des groupes sensibles aux mêmes conditions de déprotection[2].
Ensembles orthogonaux
Philip Kocienski distingue 13 ensembles orthogonaux de groupes protecteurs[2], selon qu'ils sont clivés par :
- solvolyse basique
- un acide
- les métaux lourds
- les ions fluorures
- élimination réductrice
- β-élimination
- hydrogénolyse
- oxydation
- réduction par les métaux dissous
- substitution nucléophile
- catalyse par les métaux de transition
- photolyse
- les enzymes.
Fonction protégée
Amine
Les amines présentent trois réactivités susceptibles d'être protégées[3]. Elles sont nucléophiles, basiques et, pour les amines primaires et secondaires, relativement acides. Les amines peuvent être protégées sous la forme d'imides, d'amides, de carbamates, d'imines, d'énamines, de dérivés sulfonylés, N-sulfénylés, N-alkylés ou N-silylés[4] et plus de 350 groupes protecteurs des amines ont été décrits[3].
Alcool
De très nombreuses stratégies de protection des alcools ont été développées. Cette connaissance est particulièrement importante lors d'un travail dans la série sucres[5]. Attention le groupement tosyle (Ts-) n'est pas un groupement protecteur des alcools, mais un groupement permettant leur substitution nucléophile en créant un meilleur groupe partant (pKA (acide paratoluènesulfonique/tosylate) < 0[6])[7].
| Protégé en | GP pour R-OH | Nom | Molécule | Réactif | Résistant à
A=Acide, Ox=Oxydant, Nu=Nucléophile |
Déprotection | Notes |
| Ether | Me- | Méthyl- | Me2SO4 / NaOH
MeX / base (tBuOK) |
A, Ox, Nu (Red, RM, B) | BBr3 | Déprotection difficile R-O-Me très stable | |
| Bn- | Benzyl- | PhCH2Cl / Pyr(base) | A, Ox, Nu (Red, RM, B) | H2 /Pd-C ou Na, NH3 liq | Parfois difficile à cliver
Réaction de Birch | ||
| Tr- | Trityl- | Ph3CCl / pyr (base) | Ox, Nu (H-, RM, B) | HCOOH ; H2O (Acide) | Encombrant ⇒ sélectif des alcool primaires
Carbanion stable | ||
| PMB- | p-méthoxybenzyle | PMB-Cl / base | Ox, Nu (Red, RM, B) | DDQ | Réaction radicalaire (monoélectronique) | ||
| Ether Silylé | TMS- | Triméthylsilyle | TMS-Cl + base azotée nucleophile
(Et3N, imidazole) |
Ox non acide non basique | Acide (H+,H2O)
Base (K2CO3, MeOH) HF ou ions fluorure TBAF |
Amine plus nucléophile que l'alcool + meilleur GP partant que Cl
Le Si peut être pentavalent + encombrement (Si-C : 189pm, C-C : 153pm) Catalyse Nucléophile Fragile SI-O : 530 kJ.mol-1 C-O : 340kJ.mol-1 SI-X bon Si-F 810 kJ.mol-1 C-F : 450 kJ.mol-1 | |
| TBDMS-
TBDPS- |
tert-butyldiméthylsilyle
tert-butyldiphénylsilyle |
Acide fort ou fluoré | Moins fragile car plus encombré
Non orthogonaux (choix prix ?) | ||||
| TIPS- | triisopropylsilyle | Acide fort ou fluoré | |||||
| Acetal | THP- | Tétrahydropyrane | DHP + acide (APTS) | Ox, Nu (Red, RM, B) | Acétal : BF3 (acide de Lewis)
ou acide de Brönsted (H2SO4) |
Forme un carbone asymétrique (signaux complexifiés)
Très utilisé car peu coûteux et facile à utiliser | |
| MOM-
MEM- |
Méthoxyméthyl-éther
(2-méthoxyéthoxy)méthyle éther |
MOM-CL + base | Ox, Nu (Red, RM, B) | Acide (Acétal) concentré (HCl) | Acétal = assez stable | ||
| Ester | Ac- | Acétate | Ac2O / Pyr (DMAP) ou AcCl | Acide, Ox | K2CO3,MeOH ou KOH,THF | ||
| Piv- ou Pv- | pivaloyle | PivCl / Et3N | ? | K2CO3,MeOH ou KOH,THF | |||
| Diols
(Acetal cyclique) |
1,3 dioxolane | 1,2 diol ⇒ cetone | Ox, Nu (Red, RM, B) | Acide de Lewis, HCl, etc.
Acide |
Conditions de protection sélectives
Possibilité de transacétalisation (Méthanol sous produit → colonne de distillation) | ||
| 1,3 dioxane | 1,3 diol ⇒ aldéhyde | Ox, Nu (Red, RM, B) |
Cétone
Cette fonction est protégée principalement en y ajoutant un diol (Le plus souvent l'éthane-1,2-diol) ce qui permet de former une fonction cétal (>C(OR)2). Pour réaliser la réaction inverse, il suffit de rajouter de l'eau en milieu acide pour retrouver votre fonction cétone.
Alcyne
Les alcynes peuvent réagir par leur triple liaison ou, dans le cas des alcynes terminaux, en tant qu'Acide de Brønsted.
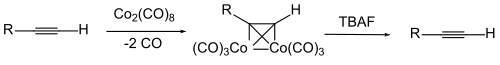
La protection de la triple liaison peut être effectuée en formant un complexe entre l'alcyne et l'octacarbonyle de dicobalt. L'alcyne est régénéré par oxydation du complexe[8],[9],[10],[11],[12].
La protection de l'acidité des alcynes terminaux est classiquement effectuée par déprotonation suivie d'une réaction avec le chlorure de triméthylsilane pour former un triméthylsilyle terminal[13]. La déprotection s'effectue par hydrolyse (carbonate de potassium et méthanol) ou par ajout d'ions fluorures, par exemple avec le TBAF[14].
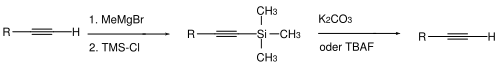
D'autres groupes trialkylsilyles peuvent être utilisés, comme le triéthylsilyle (TES), le tert-butyldiméthylsilyle (TBDMS) ou le benzyldiméthylsilyle (BDMS)[15]. Lorsque le groupe protecteur est suffisamment encombrant, la triple liaison de l'alcyne peut être protégée de l'hydrogénation catalytique sélectivement par rapport aux alcènes[16].
La protection d'un alcyne terminal peut également être effectué par un hydroxyalkyle, la déprotection peut être réalisée par un reflux dans une solution benzénique de soude[17].
Économie d'atome
Notes et références
Notes
- « orthogonal sets »
Références
- (de) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en allemand intitulé « Schutzgruppe » (voir la liste des auteurs).
- Jacques Drouin, Introduction à la chimie organique : Les molécules organiques dans votre environnement. Usages, toxicité, synthèse et réactivité, Librairie du Cèdre, 1re éd. (ISBN 2-916346-00-7), p. XVI
- (en) Philip Kocienski, Protecting groups, Stuttgart, Thieme, , 3e éd., 679 p. (ISBN 3-13-137003-3), p. 3-19
- (en) Philip Kocienski, Protecting groups, Stuttgart, Thieme, , 3e éd., 679 p. (ISBN 3-13-137003-3), p. 488
- (en) Philip Kocienski, Protecting groups, Stuttgart, Thieme, , 3e éd., 679 p. (ISBN 3-13-137003-3), p. 487
- Nicolas Rabasso, « Etude de la protection des fonctions alcool, carbonyle et amine en chimie organique », sur perso.numericable.fr (consulté le )
- J. Peter Guthrie, « Hydrolysis of esters of oxy acids: pKa values for strong acids; Brønsted relationship for attack of water at methyl; free energies of hydrolysis of esters of oxy acids; and a linear relationship between free energy of hydrolysis and pKa holding over a range of 20 pK units », Canadian Journal of Chemistry, vol. 56, no 17, , p. 2342–2354 (ISSN 0008-4042, DOI 10.1139/v78-385, lire en ligne, consulté le )
- Nicolas Rabasso, « Etude des dérivés halogénés en chimie organique », sur perso.numericable.fr (consulté le )
- (en) Barry J. Teobald, « The Nicholas reaction: the use of dicobalt hexacarbonyl-stabilised propargylic cations in synthesis », Tetrahedron, vol. 58, no 21, , p. 4133-4170 (DOI 10.1016/S0040-4020(02)00315-0)
- (en) K.M. Nicholas et R. Pettit, « An alkyne protecting group », Tetrahedron Letters, vol. 12, no 37, , p. 3475-3478 (DOI 10.1016/S0040-4039(01)97209-0)
- (en) Richard E. Connor et Kenneth M. Nicholas, « Isolation, characterization, and stability of α-[(ethynyl)dicobalt hexacarbonyl] carbonium ions », Journal of Organometallic Chemistry, vol. 125, no 2, , C45-C48 (DOI 10.1016/S0022-328X(00)89454-1)
- (en) Rosa F. Lockwood et Kenneth M. Nicholas, « Transition metal-stabilized carbenium ions as synthetic intermediates. I. α-[(alkynyl)dicobalt hexacarbonyl] carbenium ions as propargylating agents », Tetrahedron Letters, vol. 18, no 48, , p. 4163-4165 (DOI 10.1016/S0040-4039(01)83455-9)
- (en) K.M. Nicholas et R. Pettit, « On the stability of α-(alkynyl)dicobalt hexacarbonyl carbonium ions », Journal of Organometallic Chemistry, vol. 44, no 1, , C21-C24 (DOI 10.1016/0022-328X(72)80037-8)
- Jacques Drouin, Introduction à la chimie organique : Les molécules organiques dans votre environnement. Usages, toxicité, synthèse et réactivité, Librairie du Cèdre, 1re éd. (ISBN 2-916346-00-7), p. 277
- (en) Wenzel E. Davidsohn et Malcolm C. Henry, « Organometallic Acetylenes of the Main Groups III-V », Chemical Reviews, vol. 67, no 1, , p. 73-106 (DOI 10.1021/cr60245a003)
- (en) Peter G. M. Wuts et Theodora W. Greene, Protective groups : in organic synthesis, Hoboken, Wiley, , 4e éd., 1082 p. (ISBN 978-0-471-69754-1), p. 930-931
- (en) Peter G. M. Wuts et Theodora W. Greene, Protective groups : in organic synthesis, Hoboken, Wiley, , 4e éd., 1082 p. (ISBN 978-0-471-69754-1), p. 927
- (en) Peter G. M. Wuts et Theodora W. Greene, Protective groups : in organic synthesis, Hoboken, Wiley, , 4e éd., 1082 p. (ISBN 978-0-471-69754-1), p. 932
 Portail de la chimie
Portail de la chimie