Polykystose rénale autosomique dominante
La polykystose rénale autosomique dominante (PKD), aussi appelée maladie rénale polykystique ou encore Rein polykystique autosomique dominant est la plus fréquente des maladies héréditaires monogéniques du rein. Elle se caractérise par l’apparition lente et progressive de kystes principalement au niveau des reins.

| Spécialité | Génétique médicale |
|---|
| CIM-10 | Q61 |
|---|---|
| CIM-9 | 753.1 |
| OMIM | 601313 173910 |
| DiseasesDB | 10262 |
| MedlinePlus | 000502 |
| eMedicine | 376995 |
| MeSH | D016891 |
![]() Mise en garde médicale
Mise en garde médicale
Presque 10 % des individus sous rein artificiel souffrent de cette maladie. À la différence de la polykystose rénale type récessif, elle se manifeste rarement dans la période néonatale. Toutes les personnes atteintes n'évoluent pas vers l'insuffisance rénale.
Tous les autres organes peuvent être atteints comme le foie, le pancréas, les vésicules séminales et les vaisseaux sanguins. Les kystes hépatiques sont la manifestation extrarénale la plus fréquente. La rupture d'un anévrisme des artères cérébrales est une complication grave.
Les maladies proches incluent les polykystoses rénales type 1, type 2, type 3 et la maladie de Potter type 3.
Étiologie
C'est une affection de transmission autosomique dominante. Deux gènes peuvent être mutés dans la Polykystose Rénale autosomique Dominante (PKD). Dans 85 % des cas, il s'agit de PKD1 et dans 15 % des cas de PKD2.
PKD1 est localisé sur le chromosome 16 (16p13). c'est un gène de 52 Kb comportant 46 exons transcrits en un ARNm de 14 kb qui est traduit en une protéine membranaire de 4302 amino acides la polycystine-1 (PC-1). Sa fonction précise est inconnue. PKD2 est localisé sur le chromosome 4 (4q21). c'est un gène de 68 kb comportant 15 exons transcrit en un ARNm de 5 kb traduit en une protéine membranaire de 968 amino acides, la polycystine-2 (PC-2). La polycystine-2 est un canal calcique de la famille TRP.
Les deux protéines s'associent pour moduler de nombreuses voies de signalisation (JAK/STAT, Protéines G hétérotrimériques, c-Jun, ERK, AP-1, Id2, p21, etc). Elles jouent un rôle dans le contrôle de la prolifération cellulaire et dans le maintien de la polarité cellulaire. Leur expression est ubiquitaire. Elles sont toutes les deux localisées dans le cil primaire. Leur présence à ce niveau dans la cellule épithéliale rénale permet probablement de sentir le flux urinaire. La polykystose rénale autosomique dominante est donc une ciliopathie.
Il existe une grande variabilité dans l'expression clinique de la PKD. Cette variabilité est génétiquement contrôlée. Les mutations sont privées : il n'y a pas de mutations récurrentes. Les patients mutés dans PKD1 ont un âge moyen de mise en dialyse de 54 ans contre 74 ans pour ceux mutés dans PKD2[1]. Le siège de la mutation dans PKD1 a aussi un impact sur la sévérité de la maladie rénale et extra rénale.
Les kystes se développent aux dépens de 1 à 2 % des néphrons alors que toutes les cellules rénales portent la mutation de PKD1 ou PKD2. Le développement des kystes est secondaire à l'apparition d'un deuxième coup moléculaire (mutation somatique) dans le gène PKD1 ou PKD2. L'apparition des kystes dépend de cet événement. Ce modèle permet de comprendre la variabilité d'expression de la maladie. La fréquence de survenue des kystes dépend de la fréquence des mutations somatiques dans les cellules rénales. La PKD peut donc être considéré comme une maladie autosomique récessive à l'échelon cellulaire.
5 à 10 % des patients portent une mutation de novo. La maladie sous sa forme homozygote semble létale in utero.
Description
La polykystose rénale autosomique dominante est une maladie systémique. Il existe des manifestations secondaires à la présence des kystes ou d'une maladie du tissu élastique et du muscle lisse, avec atteintes rénales et extrarénales. Les deux complications majeures sont le développement de l'insuffisance rénale chronique et les anévrismes des artères cérébrales. Le diagnostic peut être posé devant une de ces manifestations cliniques, mais de plus en plus lors du dépistage familial.
Manifestations rénales
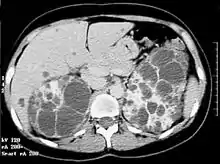
Les dilatations kystiques atteignent à la fois les canalicules biliaires et les néphrons. Les complications rénales de cette polykystose sont liées au développement, à la désorganisation, et à la destruction du parenchyme rénal normal par les kystes rénaux. La sévérité de l'atteinte rénale est directement corrélée au volume des reins. L’aspect « in utero » est rare et ressemble souvent à une polykystose rénale type récessif mais d’apparition est beaucoup plus tardive (vers 28, 32 semaines) avec absence d’anomalie du liquide amniotique. Une échographie rénale des parents est une aide pour le diagnostic. Tous les individus porteurs d'une mutation dans le gène PKD1 ou PKD2 développeront des kystes rénaux (pour les formes liées à PKD1, l'absence de kystes rénaux après 30 ans élimine le diagnostic)
La polykystose rénale autosomique dominante a une grande variabilité de présentation. Cette variabilité est visible au sein d'une même famille. Cette variabilité s'exprime sur toutes les manifestations de la maladie. En d'autres termes la présentation clinique du parent atteint ne permet pas de prédire le devenir de l'enfant atteint. La seule manifestation clinique récurrente dans une famille est la survenue d'anévrismes des artères cérébrales. Le risque pour les membres de la fratrie de présenter une telle complication si un des membres est atteint est de 25 %.
Les anomalies fonctionnelles rénales sont souvent les premiers signes cliniques. Le plus précoce est la perte de capacité de concentration normale des urines, constatée fréquemment dès l'enfance[5], conduisant à l'apparition d'un syndrome polyuropolydipsique. Il existe d'autres anomalies modifiant la composition biochimique des urines. Elles sont responsables, en partie, de l'augmentation du risque de lithiases et de l'hypertension artérielle. Le débit sanguin rénal est également diminué, contribuant à cette dernière complication.
L’hypertension artérielle est une complication précoce qui doit être systématiquement recherchée dès l'adolescence, même en l'absence d'insuffisance rénale. Sa gravité est corrélée avec le pronostic[6]. Il s'agit actuellement d'une des rares complications traitables de la maladie. Elle serait due, en partie, à l'activation du système rénine-angiotensine[7] dont la diminution du débit sanguin rénal pourrait être une cause favorisante. Cette hypertension, si elle n'est pas traitée, augmente sensiblement le risque d'insuffisance rénale.
Les douleurs rénales sont fréquentes (plus de 60 % des adultes[8]). Elles traduisent soit une augmentation importante du volume rénal qui comprime les tissus autour du rein, soit une complication : hémorragie intrakystique (qui sont cependant souvent silencieuses), infection de kystes, lithiase. L'augmentation du volume rénal peut modifier la statique lombaire et être responsable de lumbago.
La lithiase apparaît chez 20 % des malades. Ce sont des lithiases d’acide urique ou d’oxalate de calcium. Les infections rénales et vésicales sont principalement provoquées par Escherichia coli. Le risque de cancer du rein n'est pas augmenté.
L'hématurie (présence de sang dans les urines), qu'elle soit macroscopique (urines rouges) ou microscopique (sang révélé seulement à l'analyse des urines), se voit dans près de 60 % des cas[9]. L’insuffisance rénale chronique terminale nécessitant le recours à un traitement substitutif (dialyse ou transplantation) survient chez 50 % des patients. La moitié des patients porteurs d'une mutation dans PKD1 se complique d'une insuffisance rénale terminale au-delà de 54 ans. La fréquence de cette complication s'accroît ensuite avec l'âge. Pour PKD2, la sévérité est moins grande puisque la moitié des patients sont en dialyse à 74 ans. Le pronostic de l'insuffisance rénale chronique terminale est meilleur pour les patients présentant une polykystose de type dominant que pour les autres causes. Il existe une meilleure survie aussi bien en hémodialyse qu'en transplantation rénale des polykystiques. Le degré de l'insuffisance rénale est fortement corrélé avec la taille des reins[10].
Manifestations hépatiques
Les kystes hépatiques sont fréquents et leur prévalence varie avec la méthode diagnostique utilisée (beaucoup plus fréquent à l'IRM, plus sensible, qu'à l'échographie. Ils sont le plus souvent silencieux.
Ils ne se compliquent pas d'insuffisance hépatique. La complication la plus fréquente est liée au développement d'un gros foie qui peut comprimer les organes alentour et en particulier le tube digestif. L'augmentation parfois très importante du volume hépatique est responsable de douleur ou d'une impression de lourdeur. Les autres complications des kystes sont l’infection ou l’hémorragie intrakystique.
Manifestations cardiaques et vasculaires
La complication la plus redoutable est liée à la présence d'anévrismes des artères cérébrales. Ils touchent un peu moins de 10 % des patients[11] et leur fréquence passe à 25 % en cas d'antécédents familiaux[12]. Ils peuvent être asymptomatiques ou se compliquer de rupture responsable d'accidents vasculaires cérébraux hémorragiques. L'âge de survenue moyen de la rupture est de 39 ans contre 60 ans dans la population générale. Les facteurs de risque de rupture sont le siège de la mutation dans PKD1, la taille de l'anévrisme, l'hypertension artérielle et le tabagisme. Le dépistage systématique est proposé dans les familles à haut risque. Il repose sur la réalisation d'une angioIRM ou d'un scanner cérébral. S'il existe un anévrisme il est possible de proposer un traitement préventif reposant sur une exclusion de celui-ci par voie endovasculaire ou par voie chirurgicale. Dans les familles à risque la première angioIRM doit être proposée à 20 ans et réalisée tous les 10 ans si elle est normale.
Les autres complications cardiovasculaires sont le prolapsus de la valve mitrale et l'insuffisance mitrale qui touchent 25 % des patients[13]. Ces atteintes valvulaires sont le plus souvent silencieuses et non évolutives. Le risque d'hypertrophie ventriculaire gauche (augmentation de l'épaisseur du muscle cardiaque) est particulièrement important dans la population des polykystiques, en particulier s'il existe une hypertension artérielle.
Il peut exister des anévrismes sur toutes artères viscérales de gros et moyens calibres (aorte, coronaires, artères du tube digestif) mais leur fréquence est plus rare que les localisations cérébrales, ne justifiant pas un dépistage systématique.
Autres manifestations
Des kystes peuvent être présents sur d'autres organes : vésicules séminales, pancréas... Une diverticulose du côlon serait plus fréquente chez les patients en insuffisance rénale porteurs d'une polykystose de type dominante[14].
Diagnostic
La réalisation d'une échographie rénale et d'un arbre généalogique sont les deux piliers du diagnostic.
La présence de kystes hépatiques peut aider à orienter le diagnostic. Mais l'aspect des reins est en général suffisamment évocateur. L'augmentation du volume rénal est un point capital de même que l'histoire familiale traduisant la transmission dominante du trait phénotypique kyste rénale. Il existe 10 % de cas de novo (c'est-à-dire, sans histoire familiale retrouvée).
Examens complémentaires
L'échographie abdominale permet de détecter les kystes au niveau des reins.
Des critères ont été définis pour différencier la polykystose de kystes isolés dont la fréquence augmente également avec l'âge. Pour un patient présentant une histoire familiale de polykystose rénale autosomique dominante, le diagnostic est fait en présence à l'échographie de :
- deux kystes uni- ou bilatéraux avant 30 ans ;
- deux kystes dans les deux reins entre 30 ans et 59 ans ;
- quatre kystes après 60 ans[15].
Le scanner n'est pas nécessaire en première intention, même s'il visualise parfaitement les kystes, qu'ils soient rénaux ou hépatiques. l'IRM permet de quantifier le volume rénal, ce dernier étant corrélé avec le risque évolutif[16].
L'augmentation de la taille des deux reins est, en règle générale, symétrique[17].
Génétique
Le diagnostic moléculaire peut être direct par la recherche de mutation dans les gènes PKD1 et PKD2. Le rendement par des techniques modernes de l'identification des mutations est actuellement de 70 à 80 %. Il peut être indirect par la réalisation d'une analyse de liaison si les données familiales sont informatives.
Diagnostic différentiel
Les kystes simples du reins sont d'une grande banalité chez l'homme. Leur fréquence augmente avec l'âge. Les kystes microscopiques sont présents chez plus de 50 % des hommes âgés de plus de 50 ans. En échographie leur fréquence est de 1 % avant 50 ans, 10 % entre 50 et 70 ans, 20 % après 70 ans.
| Les kystes acquis | Les kystes congénitaux |
|---|---|
| Les kystes simples du rein | La maladie de Cacchi-Ricci |
| La multikystose acquise du dialysé | La dysplasie multikystique |
| Les kystes du sinus rénal | Les diverticules pyélocaliciels |
| Les kystes multiloculaires | |
| Le kyste hydatique | |
| Les kystes de l'hypokaliémie chronique | |
| Néphropathie au lithium | |
- Les maladies kystiques rénales génétiques
| Maladie | Signes associés | Gène |
|---|---|---|
| Polykystose rénale autosomique dominante | Kystes hépatiques, anévrismes des artères cérébrales | PKD1 (16p13), PKD2(4q21) |
| Maladie de von Hippel-Lindau | Cancer du rein, hemangioblastome (rétine, SNC), kyste du pancréas, phéochromocytome | VHL(3p26) |
| Sclérose tubéreuse de bourneville | Atteinte cutanée (tumeurs bénignes, taches café au lait), épilepsie, retard mental, angiomyolipomes rénaux, astrocytome | TSC1 (9q34), TSC2 (16p13) |
| Maladie kystique de la médullaire | Hyperuricémie et goutte précoce. Petits reins | UMOD (16p12.3), MCKD1 (1q21) |
| Maladie glomérulokystique | MODY V, hypolasie rénale | HNF1b (17cen) |
| Maladie | Signes associés | Gène |
|---|---|---|
| Polykystose rénale autosomique récessive | Fibrose hépatique périportale, hypertension portale | PKHD1(6p21.1-p12) |
| Néphronophtise | Petits reins, kystes médullaire, atteinte ophtalmologique possible. Syndrome de joubert. | NPHP1 (2q13), INVS (9q31), NPHP3 (3q22), NPHP4 (1p36), NPHP5 (3q13), NPHP6 (12q21), GLIS2 (16p13.3) |
| Syndrome de bardet-Biedl | Obésité, polydactylie, hypogénitalisme, rétinite pigmentaire, retard mental | BBS1 (11q13), BBS2 (16q21), BBS3 (3p12-q13), BBS4 (15q22.3), BBS5 (2q31), BBS6 (20p12), BBS7 (4q27), BBS8 (14q32.11), BBS9 (7p14), BBS10 (12q), BBS11 (9q33.1), BBS12 (4q27) |
| Maladie | Signes associés | Gène |
|---|---|---|
| Syndrome oro-facio-digital de type I | Dysmorphie faciale, anomalies des membres, retard mental. | OFD1 (Xp22.3-p22.2) |
- Les kystes simples du foie (3 à 5 %)
- Les autres maladies impliquant le rein et le foie
- Polykystose rénale type récessif
- Dysplasie rénale bilatérale isolée ou syndromique :
- Syndrome oro-facio-digital type 1
- Syndrome polydactylie-cotes courtes
- Sclérose tubéreuse de Bourneville
- Maladie de Von Hippel-Lindau
- Syndrome de Hajdu-Cheney
- Dysplasie rénale multikystique
- Maladie polykystique du foie
Traitements
Il est essentiellement symptomatique ou vise à traiter les complications de la maladie.
Lors d'une hypertension artérielle, il est logique de proposer un inhibiteur de l'enzyme de conversion ou un sartan devant le mécanisme supposé de celle-ci (activation du système rénine-angiotensine). La supériorité de ces traitements par rapport aux autres médicaments antihypertenseurs n'a cependant pas été établie de façon formelle[10]. Toutefois, il semble qu'un bon contrôle de la pression artérielle puisse ralentir l'évolution de la maladie[18].
D'autres médicaments, comme le tolvaptan[19] (un inhibiteur de la vasopressine) et l'évérolimus[20], pourraient également diminuer la progression de la polykystose.
Un kyste douloureux peut bénéficier d'une évacuation par ponction ou par chirurgie si le traitement antalgique n'est pas suffisant. L'hémorragie intrakystique est le plus souvent d'évolution simple, ne relevant que d'un simple traitement par anti-douleurs. L'infection d'un kyste est plus complexe à prendre en charge et nécessite la mise sous antibiotiques de manière parfois très prolongée, et dans quelques cas, un drainage chirurgical.
Lors d'une insuffisance rénale chronique terminale, outre la dialyse, on peut proposer une transplantation rénale, les résultats à moyen et long termes étant équivalents à ceux des autres causes d'insuffisance rénale.
En cas d'anévrisme d'une artère cérébrale, l'exclusion de ce dernier doit être discuté au cas par cas suivant sa taille et sa localisation.
Conseil génétique
La probabilité que l’enfant soit malade est de 50 %, comme pour toute maladie autosomique dominante. Un diagnostic prénatal peut être fait par la recherche de la mutation familiale si la mutation est connue, sur l'ADN fœtal prélevé par biopsie de trophoblaste ou au cours d'une amniocentèse. Il n'est, en règle, pas proposé de manière systématique, la maladie permettant une survie très prolongée et ne justifiant pas une interruption médicale de grossesse.
Perspectives
Un inhibiteur de la vasopressine, le tolvaptan, a été essayé avec un certain succès[19]. Un analogue de la somatostatine, l'octréotide, donne également des résultats prometteurs[21].
Annexes
Bibliographie
- (en) Peter C Harris, Vicente E Torres, Autosomal Dominant Polycystic Kidney Disease GeneTests: Medical Genetics Information Resource, University of Washington, Seattle. 1993-2005
Article connexe
Notes et références
- Hateboer N, van Dijk MA, Bogdanova N et als. Comparison of phenotypes of polycystic kidney disease types 1 and 2, Lancet, 1999;353:103-107
- OZ Dalgaard, Bilateral polycystic disease of the kidneys: a follow-up of two hundred and eighty-four patients and their families, Acta Med Scand 328 (1957) (suppl), pp. 1–255
- CG Iglesias, VE Torres, KP Offord, KE Holley, CM Beard and LT Kurland, Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota, Am J Kidney Dis 2 (1983), pp. 630–639
- E Higashihara, K Nutahara and M Kojima et al., Prevalence and renal prognosis of diagnosed autosomal dominant polycystic kidney disease in Japan, Nephron 80 (1998), pp. 421–427
- VE Torres, Water for ADPKD? Probably, yes, J Am Soc Nephrol, 2006;17;2089–2091
- Gabow PA, Johnson AM, Kaehny WD et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease, Kidney Int, 1992;41:1311-1319
- Chapman AB, Johnson A, Gabow PA, Schrier RW, The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease, N Engl J Med, 1990;323:1091-1096
- ZH Bajwa, KA Sial, AB Malik, TI Steinman, Pain patterns in patients with polycystic kidney disease, Kidney Int, 2004;66;1561–1569
- Grantham JJ, Autosomal dominant polycystic kidney disease, N Eng J Med, 2008;359:1477-1485
- (en) Torres VE, Harris PC, Pirson Y. « Autosomal dominant polycystic kidney disease » Lancet. 2007;369;1287-1301
- Rinkel GJ, Djibuti M, Algra A, van Gijn J, Prevalence and risk of rupture of intracranial aneurysms: a systematic review, Stroke, 1998;29:251-256
- Pirson Y, Chauveau D, Torres V, Management of cerebral aneurysms in autosomal dominant polycystic kidney disease, J Am Soc Nephrol, 2002;13:269-276
- KF Hossack, CL Leddy, AM Johnson, RW Schrier, PA Gabow, Echocardiographic findings in autosomal dominant polycystic kidney disease, N Engl J Med 1988;319;907–912
- RT Scheff, G Zuckerman, H Harter et al., Diverticular disease in patients with chronic renal failure due to polycystic kidney disease, Ann Intern Med, 1980;92;202–204
- D Ravine, RN Gibson, RG Walker, LJ Sheffield, P Kincaid-Smith, DM Danks, Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1, Lancet, 1994;343;824–827
- Chapman AB, Bost JE, Torres VE et al. Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease, Clin J Am Soc Nephrol, 2012;7:479-486
- Grantham JJ, Torres VE, Chapman AB et als. Volume progression in polycystic kidney disease, N Engl J Med, 2006;354:2122-2130
- (en) Schrier RW, Abebe KZ, Perrone RD et al. « Blood pressure in early autosomal dominant polycystic kidney disease » N Engl J Med. 2014;371:2255-2266
- (en) Torres VE, Chapman AB, Devuyst O et al. « Tolvaptan in patients with autosomal dominant polycystic kidney disease », N Engl J Med. 2012;367:2407-18.
- (en) Walz G, Budde K, Mannaa M et al. « Everolimus in patients with autosomal dominant polycystic kidney disease » N Engl J Med. 2010;363:830-840 (Erratum, N Engl J Med. 2010;363:1190)
- (en) P Ruggenenti, A Remuzzi, P Ondei et al. « Safety and efficacy of long-acting somatostatin treatment in autosomal dominant polcysytic kidney disease » Kidney Int. 2005;68;206–216
 Portail de la médecine
Portail de la médecine