Infantile myofibromatosis
| Infantile myofibromatosis | |
|---|---|
| Other names: Myofibromas, Multicentric fibromatosis,[1] Congenital generalized fibromatosis,[2] and Congenital multicentric fibromatosis[2] | |
| Specialty | Neonatology, Pediatrics |
| Usual onset | Infancy, early childhood |
| Types | Solitary tumor, Multiple tumors (no viscera involvement), and Multiple tumors (with viscera involvement) |
| Causes | Familial and non-familial somatic mutations in the PDGFRB gene |
| Prognosis | Guarded in multiple tumors (with visceral involvement); otherwise, good to excellent |
| Frequency | rare |
Infantile myofibromatosis (IMF) is a rare tumor found in 1 in 150,000 to 1 in 400,000 live births. It is nonetheless the most common tumor derived from fibrous connective tissue that occurs primarily in infants and young children. IMF tumors are benign in the sense that they do not metastasize to distant tissues although when occurring in the viscera, i.e. internal organs, carry guarded to poor prognoses and can be life-threatening, particularly in newborns and young infants.[1]
IMF tumors occur in three clinical patterns: 1) solitary IMF tumors (also called myofibromas) which often regress spontaneously and rarely cause serious issues; 2) multiple tumors (no viscera involvement) which consists of numerous (i.e. dozens to >100) IMF lesions most of which are located in the skin, subcutaneous tissues, and non-visceral but not visceral organs, may regress spontaneously, and rarely cause serious issues; and 3) multiple tumors (with viscera involvement) (also called generalized myofibromatosis) which rarely regress spontaneously[1] and consist of numerous IMF lesions in non-visceral tissues plus one or more visceral tumors that may be life-threatening.[3][4]
A minority of infantile myofibromatosis tumors present in individuals with a strong family history of the disease. These familial cases are associated with mutations in either the PDGFRB or NOTCH3 gene.[5] However, most IMF cases have no family history of the disease but nonetheless have PDGFRB gene mutations in their tumor cells; these mutations are similar to those occurring in the familial PDGFRB gene mutations.[5] Regardless of these genetic variations, all IMF tumors consist of bland-appearing, benign (i.e. non-malignant) myofibroblasts, i.e. cells that blend a variable set of features seen in fibroblasts, the most common cell type in connective tissue, with a variable set of features seen in smooth muscle cells.[3]
Treatment of IMF tumors depends upon the tumor numbers, locations, and genetic abnormalities found in each individual and often include more than one therapeutic regimen. Individuals with solitary tumors are usually treated by observation with the expectation that many of these tumors will regress spontaneiously.[6] Single tumors located in vital areas (e.g. intracranial tumors) and tumors that do not regress over suitable observation periods are often treated by surgical removal. Multiple tumors (with viscera involvement) and surgically inaccessible life-threatening IMF tumors have been treated with one or a combination of chemotherapy drugs, radiation therapy or, in tumors with certain PDGFRB gene mutations, drugs directed specifically against this mutated gene's protein product.[1]
Signs and symptoms
IMF tumors are usually painless, well-encapsulated, rubbery to hard, and freely movable-to-fixed masses.[7] They may be evident at birth in up to 60% of cases[3] but generally go undetected until they [8]are diagnosed in the first year of life,[7] uncommonly in older infants and young (<10 years/old) children,[3] or rarely in older children and adults (one individual was diagnosed with IMF at 85 years of age).[9] The tumors may be flesh-colored papules or nodules; plaques (i.e. flat-topped papules that are equal to or greater than 10 mm in largest diameter); pedunculated, calcified masses; or ulcerated, necrotic masses.[3] Most of these tumors are <1 cm in diameter[3] but can be much larger, e.g. 5.2 cm in diameter.[1] At least 9 IMF cases described in the English literature were diagnosed in fetuses based on the findings of fetal spectrogram and/or magnetic resonance imaging.[6][10] IMF tumors in the viscera or deep tissues may present with serious or life-threatening signs and symptoms of organ and/or deep tissue damage due to these tumors space-filling and pressuring effects.[8]
Some individuals with IMF present with a family history of IMF tumors. To date (2021), 20 families have been reported to carry various germline (i.e. inherited) mutations in one of their two PDGFRB genes. All of these mutations were gain of function mutations (i.e. the mutant gene's product is overactive) and inherited in an autosomal dominant manner with incomplete penetrance. Among 36 identified familial carriers of these mutant genes, 4 had no history of IMF, 3 had developed a solitary IMF, 29 had developed multiple tumors (no viscera involvement) or multiple tumors (with viscera involvement) IMF, and 2 had died. Most of these individuals were diagnosed with IMF in their infancy or early childhood.[5] Subsequent studies have found that up to 70%[11] of individuals with IMF and no family history of the disease also carry mutations in one of their two PDGFRB genes in the cells of their tumor tissues. These mutations are somatic mutations (i.e. non-inherited mutations developing only after conception) that are identical or similar to those seen in familial cases.[3][12] The familial and non-familial mutated PDGFRB genes produce highly overactive platelet-derived growth factor receptor beta (i.e. PDGFβ receptor} proteins that contribute to the development and/or progression of IMF.[13][14]
A mutation in one of the two NOTCH3 genes has been reported to occur in a single family with autosomal dominant inheritance of IMF:[1] 9 of this family's members had IMF plus the mutation while 7 of its members did not have IMF or the mutation.[15] Since the mutated NOTCH3 gene in this family produces an overactive neurogenic locus notch homolog protein 3 product that in cell culture studies increases the expression of the PDGFRB gene's product, the PDGFβ receptor, this NOTCH3 mutation may promote the development and/or progression of IMF in a manner similar to PDGFRB mutant genes, i.e. by increasing the number and therefore overall activity of the PDGFβ receptor.[15] Further studies are needed to define NOTCH3 gene mutation's frequency of occurrence in, and contribution to the development of, IMF.[5]
Solitary IMF tumors
Solitary IMF tumors represent 50 to 74% of all IMF cases.[5] These tumors commonly present with a mass that infiltrates the surrounding tissue in the head and neck areas (e.g. in the ethmoid sinus/maxillary sinus, infratemporal fossa, tongue, hard palate, behind an ear, or temporal bone); less commonly in other sites such as the scapula/axilla area, lower back, or soft tissue/bone in the lower leg; and also less commonly in visceral organs such as the pancreas,[1] spleen, lung,[6] or heart.[1] Solitary IMF tumors often (i.e. 25 to 61% of cases in different studies[6]) regress spontaneously.[16]
Multiple tumors (no viscera involvement)
Multiple tumors (no viscera involvement) typically occur as dozens to hundreds of mostly small tumors located in various areas such as the skin, muscles, and frontal eye socket,[7] These tumors do not involve visceral organs, often take a benign course, and may regress spontaneously.[3]
Multiple tumors (with viscera involvement)
Multiple tumors (with viscera involvement) typically occur as dozens to hundreds of tumors located in the same sites as multiple tumors (no viscera involvement) but also occur in one or more visceral organs such as those in the gastrointestinal tract and upper respiratory tract and/or the heart,[3] liver, cerebellum,[1] rectum,[4] parietal cortex of brain,[17] and lung.[6] This form of IMF, particularly in cases with tumors involving the gastrointestinal tract, heart, and/or lung,[5] has a far higher morbidity and mortality than the other forms[7] with death occurring in infants during the first weeks to 4 months of life[3] in 30–70% of cases[8] Death is typically due to a tumor's compression of vital organs or tissues (e.g. blood vessels).[8] However, there are uncommon cases of multiple tumors (with viscera involvement) that were not associated with serious symptoms, were observed without treatment, and over time showed spontaneous regressions of all or almost all of their tumors.[1]
Gene abnormalities
Familial IMF
Twenty families with inherited IMF have been shown to have mutations in one of their two PDGFRB genes. The PDGFRB gene is located in band 32 on the long (or "q") arm of chromosome 5.[18] Of the 20 families with PDGFRB gene mutations, 15 carried a p.Arg561Cys mutation. This mutation substitutes the amino acid Arg, i.e. arginine, for the amino acid cystine at the 561st amino acid position as numbered starting at the N-terminus of the genes product protein, the PDGFβ receptor. Four of the remaining families carried either a p.Pro560Leu (Pro=proline, Leu=leucine); p.Arg561Ser (Ser=serine); p.Lys567Glu (Lys=lysine, Glu=glutamate); or p.Pro660Thr (Thr=threonine) mutation in this protein. The NOTCH3 gene (located in band 13.12 on the short (or "p") arm of chromosome 19[19]) mutation in the carrier family was p.Leu1519Pro. The p.Arg561Cys, p.Pro560Leu, p.Arg561Ser, and p.Lys567Glu mutations are regarded as contributing to the development and/or progression of IMF while the p.Pro660Thr PDGFRB gene mutation and the p.Leu1519Pro NOTCH3 p.Leu1519Pro gene mutation require further study to determine more clearly their clinical significances.[5]
Non-familial IMF
Somatic PDGFRB gene mutations have been found in 29% of individuals with solitary IMF tumors and 68% of individuals with multiple tumors (with viscera involvement) plus multiple tumors (no visceral involvement) IMF. In about 50% of these cases, these somatic mutations were the same as those found in familial IMF with the remaining cases having mutations in other sites of the gene.[5] These other somatic mutation sites included 2 cases of p.Asp850Val (Asp=aspartate, Val=valine), 2 case of p.Asn666Lys (Asn=asparagine, Lys=lysine), and concurrent double mutations p.Arg561Cyt plus Asp666Lys in one case and p.Trp566Arg (Trp=tryptophan) plus AspN666Lys in another case. Three cases had more complex mutations involving multiple gene sites with two of them having the p.Asn666Lys mutation gene in combination with other mutations.[8]
PDGFRB gene
The PDGFRB gene product, the PDGFβ receptor, is normally highly expressed in fibroblasts[20] and other cells of mesenchymal origin including vascular smooth muscle cells.[8] When activated, the PDGFβ receptor's tyrosine kinase stimulates cell signaling proteins such as phosphatidylinositol-3 kinase, STAT proteins, phospholipase Cγ, and GRB2 which in turn promote their parent cells to grow, proliferate, and survive for abnormally prolonged times. The PDGFβ receptor gene is a proto-oncogene, i.e. a normal gene that when mutated could form a cancer-causing oncoprotein.[8] Studies show that the PDGFRB mutant proteins in IMF are constitutively overactive and that the tyrosine kinase inhibitor imatinib blocks the activity of the p.Arg561Cys and p.Asn666Lys mutants as well as the p.Arg561Cyt plus Asp666Lys and p.Trp566Arg plus AspN666Lys double mutants but not the p.Asp850Val mutant. Two other tyrosine kinase inhibitors, dasatinib and ponatinib, do inhibit the p.Asp850Val mutant's tyrosine kinase activity.[8] Limited initial studies suggest that these inhibitors are useful in treating severe cases of IMF.[8]
Pathology
Microscopic histopathologic analyses of IMF tumor tissues commonly show repeated patterns of a central zone consisting of spindle-shaped cells lining capillaries and round-to-oval cells arranged around prominent “antler-shaped” vascular spaces all of which are surrounded by a peripheral zone of clustered smooth muscle-like cells. The relative proportions of the two zones can vary within different areas of a tumor and between tumors. Immunohistochemical analyses indicate that the spindle-shaped cells strongly express smooth muscle actin, calponin, and, less commonly, desmin proteins.[3]
Diagnosis
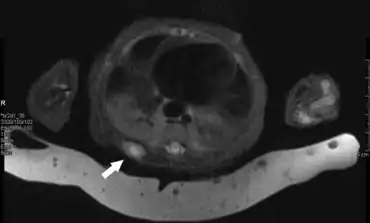
Based on their clinical presentation and gross appearance IMF tumors can be confused with a wide range of benign and malignant papular,nodular, and tumorous lesions particular in cases which involve multiple tumors. However, the presence of two zone histopathology, family history of disease, and presence of the above cited PDGFRB gene mutations are almost always definitive indicators of IMF. IMF and the classic form of mesoblastic nephroma have been suggested to be the same disease because of their very similar histopathology. However, mesoblastic nephroma tumor cells, unlike IMF tumor cells, express cyclin D1 and Beta-catenin proteins and therefore likely have a very different cellular origin than IMF tumors.[21][22] Infantile digital fibromatosis, a tumor that develops primarily in the fingers and toes, had been regarded as a type of IMF. However its tumors have a distinctly different clinical presentation and histopathology than IMF.[23] The World Health Organization (2020) redefined infantile digital fibromatosis as a benign tumor in the in the category of benign fibroblastic and myofibroblastic tumors and therefore different than IMF.[24]
Treatment
There are no controlled studies to define the optimal treatment(s) for IMF. The following give the currently widely used and recommended treatments along with the result of these treatments for the three categories of the IMF tumors.
Solitary IMF tumors
Solitary IMF tumors typically grow slowly, produce few or no symptoms, and often regress spontaneously within 18 to 24 months of diagnosis. A watchful waiting strategy is commonly used for these tumors. Visceral solitary IMF tumors that cause significant tissue injury, are located in vital areas, and/or are life threatening have been treated by surgical excision or, if surgery is deemed inappropriate, are treated with drugs and/or radiation therapy as indicated in the following section on multiple tumors (with visceral involvement).[5] Surgical excision has also be used if the diagnosis of the tumor is in doubt.[9] In rare cases, aggressive IMF tumors have recurred after regressing and required further observation and/or treatment.[5] Overall, the prognosis in almost all cases of solitary IMF is good to excellent.[3]
Multiple tumors (no viscera involvement)
Multiple tumors (no viscera involvement) also may regress spontaneously and carry a good to excellent prognosis even when they invade local tissues. However, neonates presenting with multiple IMF tumors should undergo a through examination including medical imaging tests to determine the extent of their disease, particularly with respect to determining if a visceral tumor(s) is present and therefore that the proper diagnosis is multiple tumors (with viscera involvement).[3] The treatment of multiple tumors (no viscera involvement) is similar to that used for solitary IMF tumors.[9]
Multiple tumors (with viscera involvement)
Individuals with multiple tumors (with viscera involvement) have numerous tumors one or more of which are located in and often injure a vital internal organ and may be life-threatening. Cases in which one of the tumors is in a vital organ and seriously symptomatic have been successfully treated with surgical resection. Cases where surgical resection of a single symptomatic tumor is not a good option and cases with multiple visceral tumors are treated by non-surgical methods. These tumors typically respond to conventional systemic chemotherapy with reductions in their sizes; these responses are usually observed only after several weeks of therapy.[9] Chemotherapy regimes that have successfully treated these cases include low dosage:[3] vincristine plus dactinomycin; vinblastine plus methotrexate;[5] vinblastine plus the glucocorticoid, prednisolone; vincristine plus actinomycin D plus the glucocorticoid, prednisone;[1] and vincristine plus dactinomycin plus cyclophosphamide.[4] A cytokine, interferon alpha, has also been used as a single-drug treatment in at least one of these cases.[1] Multiple tumors (with viscera involvement) have also been treated with surgical resection in combination with these drugs and/or with radiation therapy alone or combined with these drugs.[1][6]
Recently, tyrosine kinase inhibitors have been used to treat PDGFRB-associated IMF cases.[6] Imatinib or imatinib plus a second tyrosine kinase inhibitor, sorafenib, have successful treated several cases[1][5] and chemotherapy plus imatinib[4][10] have successfully treated a few cases of life-threatening IMF. Studies suggest that: 1) the tyrosine kinase inhibitor should be selected based on the PDGFRB oncoprotein's sensitivity to it (e.g. dasatinib or ponatinib should be used to treat patients with the imatinib-resistant p.Asp850Val PDGFRB gene mutation.[8]); 2) imatinib should be the initial choice and dasatinib or ponatinib be used for cases failing to respond to imatinib in cases where the PDGFRB gene mutation has not been defined;[1] and 3) the inhibitors precise dosages for use in newborns and infants are not well-defined, have serious side-affects, and therefore should only be considered for refractory, life-threatening IMF tumors.[3][8]
See also
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Manisterski M, Benish M, Levin D, Shiran SI, Sher O, Gortzak Y, Elhasid R (February 2021). "Diverse presentation and tailored treatment of infantile myofibromatosis: A single-center experience". Pediatric Blood & Cancer. 68 (2): e28769. doi:10.1002/pbc.28769. PMID 33063933. S2CID 222826034.
- 1 2 Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Fraitag S, Boccara O (August 2021). "What to Look Out for in a Newborn with Multiple Papulonodular Skin Lesions at Birth". Dermatopathology. 8 (3): 390–417. doi:10.3390/dermatopathology8030043. PMC 8395860. PMID 34449594.
- 1 2 3 4 Sparber-Sauer M, Vokuhl C, Seitz G, Sorg B, Tobias M, von Kalle T, Münter M, Bielack SS, Ladenstein R, Ljungman G, Niggli F, Frühwald M, Loff S, Klingebiel T, Koscielniak E (October 2021). "Infantile myofibromatosis: Excellent prognosis but also rare fatal progressive disease. Treatment results of five Cooperative Weichteilsarkom Studiengruppe (CWS) trials and one registry". Pediatric Blood & Cancer: e29403. doi:10.1002/pbc.29403. PMID 34636137. S2CID 238636902.
- 1 2 3 4 5 6 7 8 9 10 11 12 Hettmer S, Dachy G, Seitz G, Agaimy A, Duncan C, Jongmans M, Hirsch S, Kventsel I, Kordes U, de Krijger RR, Metzler M, Michaeli O, Nemes K, Poluha A, Ripperger T, Russo A, Smetsers S, Sparber-Sauer M, Stutz E, Bourdeaut F, Kratz CP, Demoulin JB (October 2021). "Genetic testing and surveillance in infantile myofibromatosis: a report from the SIOPE Host Genome Working Group". Familial Cancer. 20 (4): 327–336. doi:10.1007/s10689-020-00204-2. PMC 8484085. PMID 32888134.
- 1 2 3 4 5 6 7 Pekar-Zlotin M, Levinsohn-Tavor O, Livneh A, Sher O, Melcer Y, Maymon R (October 2019). "Gynecology and Oncology Fetal Myofibromatosis: A Challenge for Prenatal Diagnosis Mini Review of the English Literature". Obstetrical & Gynecological Survey. 74 (10): 607–610. doi:10.1097/OGX.0000000000000717. PMID 31670833. S2CID 204966175.
- 1 2 3 4 Lavie JL, Rogers CL, Stalder MW, St Hilaire H (January 2021). "Primary Resection and Immediate Autologous Reconstruction of Fronto-orbital Infantile Myofibromatoses". Plastic and Reconstructive Surgery. Global Open. 9 (1): e3261. doi:10.1097/GOX.0000000000003261. PMC 7858576. PMID 33552804.
- 1 2 3 4 5 6 7 8 9 10 11 Arts FA, Sciot R, Brichard B, Renard M, de Rocca Serra A, Dachy G, Noël LA, Velghe AI, Galant C, Debiec-Rychter M, Van Damme A, Vikkula M, Helaers R, Limaye N, Poirel HA, Demoulin JB (May 2017). "PDGFRB gain-of-function mutations in sporadic infantile myofibromatosis". Human Molecular Genetics. 26 (10): 1801–1810. doi:10.1093/hmg/ddx081. PMID 28334876.
- 1 2 3 4 Zhao G, Zhu M, Qin C, Liu X, Zhao X (November 2020). "Infantile Myofibromatosis: 32 Patients and Review of Literature". Journal of Pediatric Hematology/Oncology. 42 (8): 495–498. doi:10.1097/MPH.0000000000001603. PMID 31764512. S2CID 208275551.
- 1 2 Proust S, Benchimol G, Fraitag S, Starck J, Giacobbi V, Pierron G, Bodemer C, Orbach D (January 2021). "Major response to imatinib and chemotherapy in a newborn patient prenatally diagnosed with generalized infantile myofibromatosis". Pediatric Blood & Cancer. 68 (1): e28576. doi:10.1002/pbc.28576. PMID 32896962. S2CID 221540155.
- ↑ Antonescu CR, Sung YS, Zhang L, Agaram NP, Fletcher CD (May 2017). "Recurrent SRF-RELA Fusions Define a Novel Subset of Cellular Myofibroma/Myopericytoma: A Potential Diagnostic Pitfall With Sarcomas With Myogenic Differentiation". The American Journal of Surgical Pathology. 41 (5): 677–684. doi:10.1097/PAS.0000000000000811. PMC 5391281. PMID 28248815.
- ↑ Guérit E, Arts F, Dachy G, Boulouadnine B, Demoulin JB (April 2021). "PDGF receptor mutations in human diseases". Cellular and Molecular Life Sciences. 78 (8): 3867–3881. doi:10.1007/s00018-020-03753-y. PMID 33449152. S2CID 231612187.
- ↑ Wenger TL, Bly RA, Wu N, Albert CM, Park J, Shieh J, Chenbhanich J, Heike CL, Adam MP, Chang I, Sun A, Miller DE, Beck AE, Gupta D, Boos MD, Zackai EH, Everman D, Ganapathi S, Wilson M, Christodoulou J, Zarate YA, Curry C, Li D, Guimier A, Amiel J, Hakonarson H, Webster R, Bhoj EJ, Perkins JA, Dahl JP, Dobyns WB (July 2020). "Activating variants in PDGFRB result in a spectrum of disorders responsive to imatinib monotherapy". American Journal of Medical Genetics. Part A. 182 (7): 1576–1591. doi:10.1002/ajmg.a.61615. hdl:11343/275870. PMID 32500973. S2CID 219331223.
- ↑ Hassan M, Butler E, Wilson R, Roy A, Zheng Y, Liem P, Rakheja D, Pavlick D, Young LL, Rosenzweig M, Erlich R, Ali SM, Leavey PJ, Parsons DW, Skapek SX, Laetsch TW (October 2019). "Novel PDGFRB rearrangement in multifocal infantile myofibromatosis is tumorigenic and sensitive to imatinib". Cold Spring Harbor Molecular Case Studies. 5 (5): a004440. doi:10.1101/mcs.a004440. PMC 6824247. PMID 31645346.
- 1 2 Wu D, Wang S, Oliveira DV, Del Gaudio F, Vanlandewijck M, Lebouvier T, Betsholtz C, Zhao J, Jin S, Lendahl U, Karlström H (January 2021). "The infantile myofibromatosis NOTCH3 L1519P mutation leads to hyperactivated ligand-independent Notch signaling and increased PDGFRB expression". Disease Models & Mechanisms. 14 (2). doi:10.1242/dmm.046300. PMC 7927659. PMID 33509954.
- ↑ Parham DM (2018). "Fibroblastic and myofibroblastic tumors of children: new genetic entities and new ancillary testing". F1000Research. 7: 1963. doi:10.12688/f1000research.16236.1. PMC 6305242. PMID 30613391.
- ↑ Iwafuchi H, Tsuzuki T, Ito R, Miyake H, Okita H, Hamazaki M (August 2015). "Generalized infantile myofibromatosis with a monophasic primitive pattern". Pathology International. 65 (8): 432–7. doi:10.1111/pin.12312. PMID 25989870. S2CID 32983693.
- ↑ "PDGFRB platelet derived growth factor receptor beta [Homo sapiens (Human)] - Gene - NCBI". Archived from the original on 2022-01-20. Retrieved 2022-01-09.
- ↑ "NOTCH3 notch receptor 3 [Homo sapiens (Human)] - Gene - NCBI". Archived from the original on 2022-01-19. Retrieved 2022-01-09.
- ↑ Soundararajan M, Kannan S (December 2018). "Fibroblasts and mesenchymal stem cells: Two sides of the same coin?". Journal of Cellular Physiology. 233 (12): 9099–9109. doi:10.1002/jcp.26860. PMID 29943820. S2CID 49410224.
- ↑ El Demellawy D, Cundiff CA, Nasr A, Ozolek JA, Elawabdeh N, Caltharp SA, Masoudian P, Sullivan KJ, de Nanassy J, Shehata BM (2016). "Congenital mesoblastic nephroma: a study of 19 cases using immunohistochemistry and ETV6-NTRK3 fusion gene rearrangement". Pathology. 48 (1): 47–50. doi:10.1016/j.pathol.2015.11.007. PMID 27020209.
- ↑ Wang ZP, Li K, Dong KR, Xiao XM, Zheng S (2014). "Congenital mesoblastic nephroma: Clinical analysis of eight cases and a review of the literature". Oncology Letters. 8 (5): 2007–2011. doi:10.3892/ol.2014.2489. PMC 4186628. PMID 25295083.
- ↑ Laskin WB, Miettinen M, Fetsch JF (January 2009). "Infantile digital fibroma/fibromatosis: a clinicopathologic and immunohistochemical study of 69 tumors from 57 patients with long-term follow-up". The American Journal of Surgical Pathology. 33 (1): 1–13. doi:10.1097/PAS.0b013e3181788533. PMID 18830128. S2CID 30315278.
- ↑ Sbaraglia M, Bellan E, Dei Tos AP (April 2021). "The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives". Pathologica. 113 (2): 70–84. doi:10.32074/1591-951X-213. PMC 8167394. PMID 33179614.
External links
| Classification | |
|---|---|
| External resources |
|