Spinocerebellar ataxia type 6
| Spinocerebellar ataxia type 6 | |
|---|---|
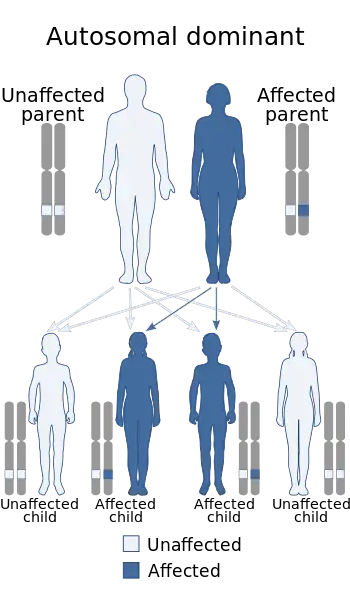 | |
| This condition is inherited in an autosomal dominant manner | |
Spinocerebellar ataxia type 6 (SCA6[1]) is a rare, late-onset, autosomal dominant disorder, which, like other types of SCA, is characterized by dysarthria, oculomotor disorders, peripheral neuropathy, and ataxia of the gait, stance, and limbs due to cerebellar dysfunction. Unlike other types, SCA 6 is not fatal. This cerebellar function is permanent and progressive, differentiating it from episodic ataxia type 2 (EA2) where said dysfunction is episodic. In some SCA6 families, some members show these classic signs of SCA6 while others show signs more similar to EA2, suggesting that there is some phenotypic overlap between the two disorders. SCA6 is caused by mutations in CACNA1A, a gene encoding a calcium channel α subunit. These mutations tend to be trinucleotide repeats of CAG, leading to the production of mutant proteins containing stretches of 20 or more consecutive glutamine residues; these proteins have an increased tendency to form intracellular agglomerations. Unlike many other polyglutamine expansion disorders expansion length is not a determining factor for the age that symptoms present.
Signs and symptoms
SCA6 is typified by progressive and permanent cerebellar dysfunction. These cerebellar signs include ataxia and dysarthria, likely caused by cerebellar atrophy. Prior to diagnosis and the onset of major symptoms, patients often report a feeling of "wooziness" and momentary imbalance when turning corners or making rapid movements. The age at which symptoms first occur varies widely, from age 19 to 71, but is typically between 43 and 52. Other major signs of SCA6 are the loss of vibratory and proprioceptive sensation and nystagmus.[2]
While most patients present with these severe progressive symptoms, others, sometimes within the same family, display episodic non-progressive symptoms more similar to episodic ataxia. Still others present with symptoms common to both SCA6 and familial hemiplegic migraine.
Pathophysiology
Most cases of SCA6 are a result of CAG repeat expansion beyond the normal range, i.e., more than 19 repeats, in the Cav2.1 calcium channel encoding gene CACNA1A.[2] This gene has two splice forms, "Q-type" and "P-type", and the polyglutamine coding CAG expansion occurs in the P-type splice form. This form is expressed heavily in the cerebellum where it is localized in Purkinje cells. In Purkinje cells from SCA6 patients, mutant Cav2.1 proteins form ovular intracellular inclusions, or aggregations, similar in many ways to those seen in other polyglutamine expansion disorders such as Huntington's disease. In cell culture models of the disease, this leads to early apoptotic cell death.[3]
Mutant channels that are able to traffic properly to the membrane have a negatively shifted voltage-dependence of inactivation. The result of this is that the channels are active for a shorter amount of time and, consequently, cell excitability is decreased.[4]
There are also a number of point mutations resulting in patients with phenotypes reminiscent of episodic ataxia and SCA6 (C271Y, G293R and R1664Q) or familial hemiplegic migraine and SCA6 (R583Q and I1710T). C287Y and G293R are both located in the pore region of domain 1 and are present in a single family each. Expression of these mutant channels results in cells with drastically decreased current density compared to wild-type expressing cells. In cell-based assays, it was found that these mutant channels aggregate in the endoplasmic reticulum, not dissimilar from that seen in the CAG expansion mutants above.[5] R1664Q is in the 4th transmembrane spanning segment of domain 4 and, presumably, affects the channel's voltage dependence of activation.[6] Little is known about the point mutations resulting in overlapping phenotypes of familial hemiplegic migraine and episodic ataxia. R583Q is present in the 4th transmembrane spanning region of domain 2 while the I1710T mutation is segment 5 of domain 4.[7][8]
Diagnosis
.jpg.webp)
Spinocerebellar Ataxia diagnosis is done via genetic testing. Your Neurologist can administer the test. Spinocerebellar Ataxia is often misdiagnosed as other diseases such as ALC or Parkinson's Disease.
Screening
There is no known prevention of spinocerebellar ataxia. Those who are believed to be at risk can have genetic sequencing of known SCA loci performed to confirm inheritance of the disorder.
Treatment
There are no drug based treatments currently available for SCA Type 6, however, there are supportive treatments that may be useful in managing symptoms. Physical Therapy, Speech Pathology can help patients manage the symptoms.
Epidemiology
The prevalence of SCA6 varies by culture. In Germany, SCA6 accounts for 10-25% of all autosomal dominant cases of SCA (SCA itself having a prevalence of 1 in 100,000).[9][10]
This prevalence in lower in Japan, however, where SCA6 accounts for only ~6% of spinocerebellar ataxias.[11]
In Australia, SCA6 accounts for 30% of spinocerebellar ataxia cases while 11% in the Dutch.[12][13]
See also
References
- ↑ "Spinocerebellar ataxia type 6: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 9 February 2023. Retrieved 19 October 2023.
- 1 2 Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton D, Amos C, Dobyns W, Subramony S, Zoghbi H, Lee C (1997). "Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel". Nat Genet. 15 (1): 62–9. doi:10.1038/ng0197-62. PMID 8988170. S2CID 9116828.
- ↑ Ishikawa K, Fujigasaki H, Saegusa H, Ohwada K, Fujita T, Iwamoto H, Komatsuzaki Y, Toru S, Toriyama H, Watanabe M, Ohkoshi N, Shoji S, Kanazawa I, Tanabe T, Mizusawa H (1999). "Abundant expression and cytoplasmic aggregations of [alpha]1A voltage-dependent calcium channel protein associated with neurodegeneration in spinocerebellar ataxia type 6". Hum Mol Genet. 8 (7): 1185–93. doi:10.1093/hmg/8.7.1185. PMID 10369863.
- ↑ Toru S, Murakoshi T, Ishikawa K, Saegusa H, Fujigasaki H, Uchihara T, Nagayama S, Osanai M, Mizusawa H, Tanabe T (2000). "Spinocerebellar ataxia type 6 mutation alters P-type calcium channel function". J Biol Chem. 275 (15): 10893–8. doi:10.1074/jbc.275.15.10893. PMID 10753886.
- ↑ Wan J, Khanna R, Sandusky M, Papazian D, Jen J, Baloh R (2005). "CACNA1A mutations causing episodic and progressive ataxia alter channel trafficking and kinetics". Neurology. 64 (12): 2090–7. doi:10.1212/01.WNL.0000167409.59089.C0. PMID 15985579. S2CID 5679518.
- ↑ Tonelli A, D'Angelo M, Salati R, Villa L, Germinasi C, Frattini T, Meola G, Turconi A, Bresolin N, Bassi M (2006). "Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene". J Neurol Sci. 241 (1–2): 13–7. doi:10.1016/j.jns.2005.10.007. PMID 16325861. S2CID 36806418.
- ↑ Alonso I, Barros J, Tuna A, Coelho J, Sequeiros J, Silveira I, Coutinho P (2003). "Phenotypes of spinocerebellar ataxia type 6 and familial hemiplegic migraine caused by a unique CACNA1A missense mutation in patients from a large family". Arch Neurol. 60 (4): 610–4. doi:10.1001/archneur.60.4.610. hdl:10400.16/349. PMID 12707077.
- ↑ Kors E, Vanmolkot K, Haan J, Kheradmand Kia S, Stroink H, Laan L, Gill D, Pascual J, van den Maagdenberg A, Frants R, Ferrari M (2004). "Alternating hemiplegia of childhood: no mutations in the second familial hemiplegic migraine gene ATP1A2". Neuropediatrics. 35 (5): 293–6. doi:10.1055/s-2004-821082. PMID 15534763.
- ↑ Riess O, Schöls L, Bottger H, Nolte D, Vieira-Saecker A, Schimming C, Kreuz F, Macek M, Krebsová A, Klockgether T, Zühlke C, Laccone F (1997). "SCA6 is caused by moderate CAG expansion in the alpha1A-voltage-dependent calcium channel gene". Hum Mol Genet. 6 (8): 1289–93. doi:10.1093/hmg/6.8.1289. PMID 9259275.
- ↑ Schöls L, Amoiridis G, Büttner T, Przuntek H, Epplen J, Riess O (1997). "Autosomal dominant cerebellar ataxia: phenotypic differences in genetically defined subtypes?". Ann Neurol. 42 (6): 924–32. doi:10.1002/ana.410420615. PMID 9403486. S2CID 32742844.
- ↑ Watanabe H, Tanaka F, Matsumoto M, Doyu M, Ando T, Mitsuma T, Sobue G (1998). "Frequency analysis of autosomal dominant cerebellar ataxias in Japanese patients and clinical characterization of spinocerebellar ataxia type 6". Clin Genet. 53 (1): 13–9. doi:10.1034/j.1399-0004.1998.531530104.x. PMID 9550356.
- ↑ Storey E, du Sart D, Shaw J, Lorentzos P, Kelly L, McKinley Gardner R, Forrest S, Biros I, Nicholson G (2000). "Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with spinocerebellar ataxia". Am J Med Genet. 95 (4): 351–7. doi:10.1002/1096-8628(20001211)95:4<351::AID-AJMG10>3.0.CO;2-R. PMID 11186889.
- ↑ Sinke R, Ippel E, Diepstraten C, Beemer F, Wokke J, van Hilten B, Knoers N, van Amstel H, Kremer H (2001). "Clinical and molecular correlations in spinocerebellar ataxia type 6: a study of 24 Dutch families". Arch Neurol. 58 (11): 1839–44. doi:10.1001/archneur.58.11.1839. PMID 11708993.
External links
| Classification | |
|---|---|
| External resources |