Usher syndrome
| Usher syndrome | |
|---|---|
| Other names: Usher–Hallgren syndrome | |
 | |
| Usher syndrome is inherited in an autosomal recessive pattern. The genes implicated in Usher syndrome are described below. | |
Usher syndrome, also known as Hallgren syndrome, Usher–Hallgren syndrome, retinitis pigmentosa–dysacusis syndrome or dystrophia retinae dysacusis syndrome,[1] is a rare genetic disorder caused by a mutation in any one of at least 11 genes resulting in a combination of hearing loss and visual impairment. It is a major cause of deafblindness and is at present incurable.
Usher syndrome is classed into three subtypes (I, II and III) according to the genes responsible and the onset of deafness. All three subtypes are caused by mutations in genes involved in the function of the inner ear and retina. These mutations are inherited in an autosomal recessive pattern.
The occurrence of Usher syndrome varies across the world and across the different syndrome types, with rates as high as 1 in 12,500 in Germany[2] to as low as 1 in 28,000 in Norway.[3] Type I is most common in Ashkenazi Jewish and Acadian populations, and type III is rarely found outside Ashkenazi Jewish and Finnish[4] populations. Usher syndrome is named after Scottish ophthalmologist Charles Usher, who examined the pathology and transmission of the syndrome in 1914.
Types
Usher syndrome I
People with Usher I are born profoundly deaf and begin to lose their vision in the first decade of life. They also exhibit balance difficulties and learn to walk slowly as children, due to problems in their vestibular system.
Usher syndrome type I can be caused by mutations in any one of several different genes: CDH23, MYO7A, PCDH15, USH1C and USH1G. These genes function in the development and maintenance of inner ear structures such as hair cells (stereocilia), which transmit sound and motion signals to the brain. Alterations in these genes can cause an inability to maintain balance (vestibular dysfunction) and hearing loss. The genes also play a role in the development and stability of the retina by influencing the structure and function of both the rod photoreceptor cells and supporting cells called the retinal pigmented epithelium. Mutations that affect the normal function of these genes can result in retinitis pigmentosa and resultant vision loss.
Worldwide, the estimated prevalence of Usher syndrome type I is 3 to 6 per 100,000 people in the general population. Type I has been found to be more common in people of Ashkenazi Jewish ancestry (central and eastern European) and in the French-Acadian populations (Louisiana).
Usher syndrome II
People with Usher II are not born deaf and are generally hard-of-hearing rather than deaf, and their hearing does not degrade over time; moreover, they do not seem to have noticeable problems with balance. They also begin to lose their vision later (in the second decade of life) and may preserve some vision even into middle age.
Usher syndrome type II may be caused by mutations in any of three different genes: USH2A, GPR98 and DFNB31. The protein encoded by the USH2A gene, usherin, is located in the supportive tissue in the inner ear and retina. Usherin is critical for the proper development and maintenance of these structures, which may help explain its role in hearing and vision loss. The location and function of the other two proteins are not yet known.
Usher syndrome type II occurs at least as frequently as type I, but because type II may be underdiagnosed or more difficult to detect, it could be up to three times as common as type I.
Usher syndrome III
People with Usher syndrome III are not born deaf but experience a progressive loss of hearing, and roughly half have balance difficulties.
Mutations in only one gene, CLRN1, have been linked to Usher syndrome type III. CLRN1 encodes clarin-1, a protein important for the development and maintenance of the inner ear and retina. However, the protein's function in these structures, and how its mutation causes hearing and vision loss, is still poorly understood.
The frequency of Usher syndrome type III is significant only in the Finnish population[4] as well as the population of Birmingham, UK,[5] and individuals of Ashkenazi Jewish heritage. It has been noted rarely in a few other ethnic groups.
Symptoms and signs
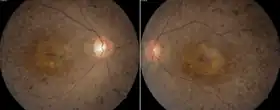
Usher syndrome is characterized by hearing loss and a gradual visual impairment. The hearing loss is caused by a defective inner ear, whereas the vision loss results from retinitis pigmentosa (RP), a degeneration of the retinal cells. Usually, the rod cells of the retina are affected first, leading to early night blindness (nyctalopia) and the gradual loss of peripheral vision. In other cases, early degeneration of the cone cells in the macula occurs, leading to a loss of central acuity. In some cases, the foveal vision is spared, leading to "doughnut vision"; central and peripheral vision are intact, but an annulus exists around the central region in which vision is impaired.
Cause
| Type | Freq[6] | Gene locus | Gene | Protein | Function | Size (AA) | UniProt | OMIM |
|---|---|---|---|---|---|---|---|---|
| USH1B | 39–55% | 11q13.5 | MYO7A | Myosin VIIA | Motor protein | 2215 | Q13402 | 276900 |
| USH1C | 6–7% | 11p15.1-p14 | USH1C | Harmonin | PDZ-domain protein | 552 | Q9Y6N9 | 276904 |
| USH1D | 19–35% | 10q21-q22 | CDH23 | Cadherin 23 | Cell adhesion | 3354 | Q9H251 | 601067 |
| USH1E | rare | 21q21 | ? | ? | ? | ? | ? | 602097 |
| USH1F | 11–19% | 10q11.2-q21 | PCDH15 | Protocadherin 15 | Cell adhesion | 1955 | Q96QU1 | 602083 |
| USH1G | 7% | 17q24-q25 | USH1G | SANS | Scaffold protein | 461 | Q495M9 | 606943 |
| USH2A | 80% | 1q41 | USH2A | Usherin | Transmembrane linkage | 5202 | O75445 | 276901 |
| USH2C | 15% | 5q14.3-q21.1 | GPR98 | VLGR1b | Very large GPCR | 6307 | Q8WXG9 | 605472 |
| USH2D | 5% | 9q32-q34 | DFNB31 | Whirlin | PDZ-domain protein | 907 | Q9P202 | 611383 |
| USH3A | 100% | 3q21-q25 | CLRN1 | Clarin-1 | Synaptic shaping | 232 | P58418 | 276902 |
Usher syndrome is inherited in an autosomal recessive pattern. Several genes have been associated with Usher syndrome using linkage analysis of patient families (Table 1) and DNA sequencing of the identified loci.[7][8] A mutation in any one of these genes is likely to result in Usher syndrome.
The clinical subtypes Usher I and II are associated with mutations in any one of six (USH1B-G) and three (USH2A ,C-D) genes, respectively, whereas only one gene, USH3A, has been linked to Usher III so far. Two other genes, USH1A and USH2B, were initially associated with Usher syndrome, but USH2B has not been verified and USH1A was incorrectly determined and does not exist.[9] Research in this area is ongoing.
Using interaction analysis techniques, the identified gene products could be shown to interact with one another in one or more larger protein complexes. If one of the components is missing, this protein complex cannot fulfil its function in the living cell, and it probably comes to the degeneration the same. The function of this protein complex has been suggested to participate in the signal transduction or in the cell adhesion of sensory cells.[8]
A study shows that three proteins related to Usher syndrome genes (PCDH15, CDH23, GPR98) are also involved in auditory cortex development, in mouse and macaque. Their lack of expression induces a decrease in the number of parvalbumin interneurons. Patients with mutations for these genes could have consequently auditory cortex defects.[10]
Pathophysiology
The progressive blindness of Usher syndrome results from retinitis pigmentosa.[11][12] The photoreceptor cells usually start to degenerate from the outer periphery to the center of the retina, including the macula. The degeneration is usually first noticed as night blindness (nyctalopia); peripheral vision is gradually lost, restricting the visual field (tunnel vision), which generally progresses to complete blindness. The qualifier pigmentosa reflects the fact that clumps of pigment may be visible by an ophthalmoscope in advanced stages of degeneration.[13]
The hearing impairment associated with Usher syndrome is caused by damaged hair cells in the cochlea of the inner ear inhibiting electrical impulses from reaching the brain. It is a form of dysacusis.
Diagnosis
Since Usher syndrome is incurable at present, it is helpful to diagnose children well before they develop the characteristic night blindness. Some preliminary studies have suggested as many as 10% of congenitally deaf children may have Usher syndrome.[1] However, a misdiagnosis can have bad consequences.
The simplest approach to diagnosing Usher syndrome is to test for the characteristic chromosomal mutations. An alternative approach is electroretinography, although this is often disfavored for children, since its discomfort can also make the results unreliable.[1] Parental consanguinity is a significant factor in diagnosis. Usher syndrome I may be indicated if the child is profoundly deaf from birth and especially slow in walking.
Thirteen other syndromes may exhibit signs similar to Usher syndrome, including Alport syndrome, Alström syndrome, Bardet–Biedl syndrome, Cockayne syndrome, spondyloepiphyseal dysplasia congenita, Flynn–Aird syndrome, Friedreich ataxia, Hurler syndrome (MPS-1), Kearns–Sayre syndrome (CPEO), Norrie syndrome, osteopetrosis (Albers–Schonberg disease), Refsum disease (phytanic acid storage disease) and Zellweger syndrome (cerebrohepatorenal syndrome).
Classification
Although Usher syndrome has been classified clinically in several ways,[14][12][15] the prevailing approach is to classify it into three clinical sub-types called Usher I, II and III in order of decreasing severity of deafness.[11][13] Although it was previously believed that there was an Usher syndrome type IV, researchers at the University of Iowa recently confirmed that there is no USH type IV. As described below, these clinical subtypes may be further subdivided by the particular gene mutated; people with Usher I and II may have any one of six and three genes mutated, respectively, whereas only one gene has been associated with Usher III. The function of these genes is still poorly understood.
Usher syndrome is a variable condition; the degree of severity is not tightly linked to whether it is Usher I, II or III. For example, someone with type III may be unaffected in childhood but go on to develop a profound hearing loss and a very significant loss of sight by early-to-mid adulthood. Similarly, someone with type I, who is therefore profoundly deaf from birth, may keep good central vision until the sixth decade of life or even beyond. People with type II, who have useful hearing with a hearing aid, can experience a wide range of severity of the RP. Some may maintain good reading vision into their 60s, while others cannot see to read while still in their 40s.
Since Usher syndrome is inherited in an autosomal recessive pattern, both males and females are equally likely to inherit it. Consanguinity of the parents is a risk factor.
Treatment
Since Usher syndrome results from the loss of a gene, gene therapy that adds the proper protein back ("gene replacement") may alleviate it, provided the added protein becomes functional. Recent studies of mouse models have shown one form of the disease—that associated with a mutation in myosin VIIa—can be alleviated by replacing the mutant gene using a lentivirus.[16] However, some of the mutated genes associated with Usher syndrome encode very large proteins—most notably, the USH2A and GPR98 proteins, which have roughly 6000 amino-acid residues. Gene replacement therapy for such large proteins may be difficult.
Epidemiology
Usher syndrome is responsible for the majority of deafblindness.[17] It occurs in roughly 1 in 23,000 people in the United States,[18] 1 in 28,000 in Norway,[3] and 1 in 12,500 in Germany.[2] People with Usher syndrome represent roughly one-sixth of people with retinitis pigmentosa.[13]
History
Usher syndrome is named after the Scottish ophthalmologist Charles Usher, who examined the pathology and transmission of this illness in 1914 on the basis of 69 cases.[19] However, it was first described in 1858 by Albrecht von Gräfe, a pioneer of modern ophthalmology.[20] He reported the case of a deaf patient with retinitis pigmentosa, who had two brothers with the same symptoms. Three years later, one of his students, Richard Liebreich, examined the population of Berlin for disease pattern of deafness with retinitis pigmentosa.[21] Liebreich noted Usher syndrome to be recessive, since the cases of blind-deafness combinations occurred particularly in the siblings of blood-related marriages or in families with patients in different generations. His observations supplied the first proofs for the coupled transmission of blindness and deafness, since no isolated cases of either could be found in the family trees.
Animal models of this human disease (such as knockout mice and zebrafish) have been developed recently to study the effects of these gene mutations and to test potential cures for Usher syndrome.
Notable cases
- Rebecca Alexander, a psychotherapist, author, and recipient of the Helen Keller Achievement Award.
- Christine "Coco" Roschaert, director of the Nepal Deafblind Project, kick-off speaker for Deaf Awareness Week at the University of Vermont, and participant in the Gallaudet United Now Movement.[22]
- Catherine Fischer wrote her autobiography of growing up with Usher syndrome in Louisiana, entitled Orchid of the Bayou.[23]
- Vendon Wright has written two books describing his life with Usher syndrome, I was blind but now I can see[24] and Through my eyes.[25]
- Christian Markovic, and blind-deaf illustrator and designer; Fuzzy Wuzzy Designs.[26]
- John Tracy, the son of actor Spencer Tracy and namesake of the oralist John Tracy Clinic.
- James D. Watson, DNA helix co-discoverer and Nobel laureate, has homozygous USH1B mutations, according to his published genome.[27] It is not clear why he did not develop the syndrome. This lack of genetic penetrance argues that expression of the phenotype of Usher syndrome may be more complex than originally assumed.
- The Israeli Nalaga'at (do touch) Deaf-blind Acting Ensemble consists of 11 deaf-blind actors, most of whom are diagnosed with Usher syndrome. The theater group has put on several productions and appeared both locally in Israel and abroad in London and Broadway.[28]
- Katie Kelly, a gold medal winning paraolympian.
- Teigan Van Roosmalen, paraolympian.
- Cyril Axelrod, Catholic priest.
- Rachel Chaikof, early cochlear implant recipient and founder of cochlearimplantonline.com.
- Robert Tarango, first deafblind person to star in a movie, in the role of Artie in the Oscar-nominated short film Feeling Through.
References
- 1 2 3 Mets MB, Young NM, Pass A, Lasky JB (2000). "Early diagnosis of Usher syndrome in children". Transactions of the American Ophthalmological Society. 98: 237–45. PMC 1298229. PMID 11190026.
- 1 2 Otterstedde CR, Spandau U, Blankenagel A, Kimberling WJ, Reisser C (2001). "A new clinical classification for Usher's syndrome based on a new subtype of Usher's syndrome type I". Laryngoscope. 111 (1): 84–86. doi:10.1097/00005537-200101000-00014. PMID 11192904.
- 1 2 Grøndahl J (1987). "Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway". Clin. Genet. 31 (4): 255–264. doi:10.1111/j.1399-0004.1987.tb02804.x. PMID 3594933. S2CID 26853136.
- 1 2 Pakarinen L, Tuppurainen K, Laipapala P, Mäntyjärvi M, Puhakka H (1996). "The ophthalmological course of Usher syndrome type III". International Ophthalmology. 19 (5): 307–311. doi:10.1007/BF00130927. PMID 8864816. S2CID 26501078.
- ↑ Hope CI, Bundey S, Proops D, Fielder AR (1997). "Usher syndrome in the city of Birmingham — prevalence and clinical classification". British Journal of Ophthalmology. 81 (1): 46–53. doi:10.1136/bjo.81.1.46. PMC 1721995. PMID 9135408.
- ↑ Roux AF, Faugere V, Le Guedard S, Pallares-Ruiz N, Vielle A, Chambert S, Marlin S, Hamel C, Gilbert B, Malcolm S, Claustres M (2006). "Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%". J Med Genet. 43 (9): 763–768. doi:10.1136/jmg.2006.041954. PMC 2564578. PMID 16679490.
Ouyang XM, Yan D, Du LL, Hejtmancik JF, Jacobson SG, Nance WE, Li AR, Angeli S, Kaiser M, Newton V, Brown SD, Balkany T, Liu XZ (2005). "Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population". Hum Genet. 116 (4): 292–299. doi:10.1007/s00439-004-1227-2. PMID 15660226. S2CID 22812718. - ↑ Petit, C (2001). "Usher syndrome: from genetics to pathogenesis" (PDF). Annual Review of Genomics and Human Genetics. 2: 271–97. doi:10.1146/annurev.genom.2.1.271. PMID 11701652. S2CID 505750. Archived from the original (PDF) on 2019-05-03.
- 1 2 Reiners, J; Nagel-Wolfrum, K; Jürgens, K; Märker, T; Wolfrum, U (2006). "Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease" (PDF). Experimental Eye Research. 83 (1): 97–119. doi:10.1016/j.exer.2005.11.010. PMID 16545802. Archived from the original (PDF) on 2019-05-03.
- ↑ Gerber, S; Bonneau, D; Gilbert, B; Munnich, A; Dufier, JL; Rozet, JM; Kaplan, J (2006). "USH1A: chronicle of a slow death". American Journal of Human Genetics. 78 (2): 357–9. doi:10.1086/500275. PMC 1380243. PMID 16400615.
- ↑ Libé-Philippot, Baptiste; Michel, Vincent; Monvel, Jacques Boutet de; Gal, Sébastien Le; Dupont, Typhaine; Avan, Paul; Métin, Christine; Michalski, Nicolas; Petit, Christine (2017-07-25). "Auditory cortex interneuron development requires cadherins operating hair-cell mechanoelectrical transduction". Proceedings of the National Academy of Sciences. 114 (30): 7765–7774. doi:10.1073/pnas.1703408114. ISSN 0027-8424. PMC 5544301. PMID 28705869.
- 1 2 Smith RJ, Berlin CI, Hejtmancik JF, Keats BJ, Kimberling WJ, Lewis RA, et al. (1994). "Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium". American Journal of Medical Genetics. 50 (1): 32–38. doi:10.1002/ajmg.1320500107. PMID 8160750.
- 1 2 Fishman GA, Kumar A, Joseph ME, Torok N, Andersonj RJ (1983). "Usher's syndrome: ophthalmic and neuro-otologic findings suggesting genetic heterogeneity". Archives of Ophthalmology. 101 (9): 1367–1374. doi:10.1001/archopht.1983.01040020369005. PMID 6604514.
- 1 2 3 Williams DS (2007). "Usher syndrome: Animal models, retinal function of Usher proteins, and prospects for gene therapy". Vision Research. 48 (3): 433–41. doi:10.1016/j.visres.2007.08.015. PMC 2680226. PMID 17936325.
- ↑
Hammerschlag V (1907). "Zur Kenntnis der hereditaer-degenerativen Taubstummen und ihre differential diagnostische Bedeutung". Z. Ohrenheilk. 54: 18–36.
Bell J (1933). Retinitis Pigmentosa and Allied Diseases (2nd ed.). London: Cambridge University Press.
Hallgren B (1959). "Retinitis pigmentosa combined with congenital deafness with vestibulo-cerebellar ataxia and mental abnormality in a proportion of cases: Clinical and geneto-statistical survey". Acta Psychiatr Scand Suppl. 34 (138): 9–101. doi:10.1111/j.1600-0447.1959.tb08605.x. PMID 14399116. S2CID 221393918.
Merin S, Auerbach E (1976). "Retinitis pigmentosa". Surv. Ophthalmol. 20 (5): 303–345. doi:10.1016/S0039-6257(96)90001-6. PMID 817406.
Davenport S, Omenn G (1977). The Heterogeneity of Usher Syndrome (volume 426 ed.). Amsterdam: Excerpta Medica Foundation.
Gorlin R, Tilsner T, Feinstein S, Duvall AJ (1979). "Usher syndrome type III". Arch. Otolaryngol. 105 (6): 353–354. doi:10.1001/archotol.1979.00790180051011. PMID 454290. - ↑ Sankila EM, Pakarinen H, Kääriäinen H, Aittomäki K, Karjalainen S, Sistonen P, de la Chapelle A (1995). "Assignment of Usher syndrome type III (USH3) gene to chromosome 3q". Hum. Mol. Genet. 4 (1): 93–98. doi:10.1093/hmg/4.1.93. PMID 7711740.
- ↑ Hashimoto T, Gibbs D, Lillo C, Azarian SM, Legacki E, Zhang XM, Yang XJ, Williams DS (2007). "Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B". Gene Therapy. 14 (7): 584–594. doi:10.1038/sj.gt.3302897. PMID 17268537.
- ↑ Vernon M (1969). "Usher's syndrome — deafness and progressive blindness. Clinical cases, prevention, theory and literature survey". Journal of Chronic Diseases. 22 (3): 133–151. doi:10.1016/0021-9681(69)90055-1. PMID 4897966.
- ↑ Boughman J, Vernon M, Shaver K (1983). "Usher syndrome: Definition and estimate of prevalence from two high-risk populations". Journal of Chronic Diseases. 36 (8): 595–603. doi:10.1016/0021-9681(83)90147-9. PMID 6885960.
- ↑ Usher C (1914). "On the inheritance of Retinitis pigmentosa with notes of cases". Roy. Lond. Ophthalmol. Hosp. Rep. 19: 130–236.
- ↑ von Gräfe A (1858). "Exceptionelles Verhalten des Gesichtsfeldes bei Pigmententartung der Netzhaut". Archiv für Ophthalmologie. 4: 250–253.
- ↑ Liebreich R (1861). "Abkunft aus Ehen unter Blutsverwandten als Grund von Retinitis pigmentosa". Dtsch. Klin. 13: 53.
- ↑ "Tactile The World". Tactile The World. Archived from the original on 2021-01-16. Retrieved 2021-03-29.
- ↑ Carroll C, Fischer CH (2001). Orchid of the Bayou: A Deaf Woman Faces Blindness. Gallaudet University Press. ISBN 978-1-56368-104-2.
- ↑ Wright V (2007). I was blind but now I can see. Authorhouse. ISBN 978-1-4208-9101-0.
- ↑ Wright V (2007). Through my eyes. Pipers' Ash Ltd. ISBN 978-1-904494-86-7.
- ↑ "Who's Fuzzy". Fuzzy Wuzzy Design. Archived from the original on 2021-06-29. Retrieved 2021-03-29.
- ↑ Green RC, Annas GJ (2008). "The Genetic Privacy of Presidential Candidates". New England Journal of Medicine. 359 (21): 2192–2193. doi:10.1056/NEJMp0808100. PMC 2925179. PMID 19020322.
- ↑ "Archive copy". Archived from the original on 2010-11-24. Retrieved 2021-03-29.
{{cite web}}: CS1 maint: archived copy as title (link)
Further reading
- Stiefel SH, Lewis RA (1991). The Madness of Usher's: Coping With Vision and Hearing Loss/Usher Syndrome Type II. Business of Living Publications. ISBN 978-1-879518-06-3.
- Duncan E, Prickett HT (1988). Usher's Syndrome: What It Is, How to Cope, and How to Help. Charles C. Thomas. ISBN 978-0-398-05481-6.
- Vernon M (1986). Answers to your questions about Usher's syndrome (retinitis pigmentosa with hearing loss). Foundation Fighting Blindness. ASIN B00071QLJ6.
- Vernon M (1969). Usher's syndrome: Deafness and progressive blindness : clinical cases, prevention, theory and literature survey. Pergamon Press. ASIN B0007JHOJ4.
External links
| Classification | |
|---|---|
| External resources |
|
- GeneReviews/NCBI/NIH/UW entry on Usher Syndrome Type I Archived 2017-01-18 at the Wayback Machine
- GeneReviews/NCBI/NIH/UW entry on Usher Syndrome Type II Archived 2020-11-23 at the Wayback Machine
- NCBI Genetic Testing Registry Archived 2018-12-05 at the Wayback Machine
- General overview from the NIH Archived 2021-03-24 at the Wayback Machine
- Usher Syndrome Information from the National Institute on Deafness and Other Communication Disorders (NIDCD).