داء الخلايا المنجلية
داء الخلايا المنجلية أو داء الكريات المنجلية هو مجموعة من اضطرابات الدم الموروثة عادة من والدي الشخص.[3] يعرف النوع الأكثر شيوعًا باسم فقر الدم المنجلي.[3] إنه يؤدي إلى خلل في الهيموجلوبين البروتيني الحامل للأكسجين الموجود في خلايا الدم الحمراء.[3] هذا يؤدي إلى شكل صلب يشبه المنجل في ظل ظروف معينة.[3] تبدأ مشاكل مرض الخلايا المنجلية عادة حوالي 5 إلى 6 أشهر من العمر.[2] قد تتطور عدد من المشاكل الصحية، مثل نوبات الألم (المعروفة باسم أزمة الخلايا المنجلية)، فقر الدم، تورم اليدين والقدمين، الالتهابات البكتيرية والسكتة الدماغية.[2] قد يتطور الألم طويل المدى مع تقدم الناس في السن.[3] يتراوح متوسط العمر المتوقع في العالم المتقدم بين 40 و60 عامًا.[3]
| داء الخلايا المنجلية | |
|---|---|
| Sickle cell disease | |
 الشكل (أ) يعرض خلايا الدم الحمراء الطبيعية التي تتدفق بحرية من خلال الأوردة. يظهر الشكل قطاع عريض من خلايا الدم الحمراء الطبيعية مع الهيموغلوبين الطبيعي. الشكل (ب) يظهر خلايا الدم الحمراء المنجلية غير الطبيعية، تلتصق وتتجمع عند نقطة التفرع في الوريد. وتظهر الصورة قطاع عريض من خلية المنجل مع خيوط طويلة مبلمرة تمتد وتشوه شكل الخلية. الشكل (أ) يعرض خلايا الدم الحمراء الطبيعية التي تتدفق بحرية من خلال الأوردة. يظهر الشكل قطاع عريض من خلايا الدم الحمراء الطبيعية مع الهيموغلوبين الطبيعي. الشكل (ب) يظهر خلايا الدم الحمراء المنجلية غير الطبيعية، تلتصق وتتجمع عند نقطة التفرع في الوريد. وتظهر الصورة قطاع عريض من خلية المنجل مع خيوط طويلة مبلمرة تمتد وتشوه شكل الخلية. | |
| تسميات أخرى | مرض الخلايا المنجليه |
| معلومات عامة | |
| الاختصاص | علم الدم، علم الوراثة الطبية |
| من أنواع | اضطراب صبغي جسدي متنحي ، واعتلال الهيموغلوبين، وفقر الدم الانحلالي الخلقي، ومرض |
| الأسباب | |
| الأسباب | وراثي[1] |
| المظهر السريري | |
| البداية المعتادة | 5-6 أشهر[2] |
| الأعراض | نوبات الألم، فقر الدم، تورم اليدين والقدمين، الالتهابات البكتيرية، السكتة الدماغية[2] |
| المضاعفات | ألم مزمن، سكتة دماغية، نخر لاوعائي، حصاة صفراوية، قرحة وريدية، قساح، فرط ضغط الدم الرئوي، مشاكل في الرؤية، قصور كلوي[3] |
| الإدارة | |
| التشخيص | تحليل الدم[4] |
| العلاج | التطعيم، المضادات الحيوية، تناول كميات كبيرة من السوائل، تناول حمض الفوليك، مسكنات الألم، نقل الدم[5] |
| المآل | متوسط العمر المتوقع 40-60 عامًا (الدول المتقدمة)[3] |
| الوبائيات | |
| انتشار المرض | 4.4 مليون شخص[6] |
| الوفيات | 114,800 (2015)[7] |

يحدث مرض الخلايا المنجلية عندما يرث الشخص نسختين غير طبيعيتين من جين بيتا جلوبين (HBB) الذي يصنع الهيموجلوبين، واحدة من كل والد.[1] يحدث هذا الجين في الكروموسوم 11.[8] توجد عدة أنواع فرعية، اعتمادًا على الطفرة الدقيقة في كل جين من جينات الهيموجلوبين.[3] يمكن أن ينجم الهجوم عن طريق التغيرات في درجات الحرارة، الإجهاد، الجفاف والارتفاعات العالية.[2] الشخص الذي لديه نسخة واحدة غير طبيعية ليس لديه أعراض عادة ويقال إنه يعاني من سمة الخلية المنجلية.[1] يشار إلى هؤلاء الأشخاص أيضًا باسم الحاملين.[9] يتم التشخيص عن طريق فحص الدم، تختبر بعض البلدان جميع الأطفال عند الولادة بحثًا عن المرض.[4] التشخيص ممكن أيضا أثناء الحمل.[4]
قد تشمل رعاية الأشخاص المصابين بمرض الخلايا المنجلية الوقاية من العدوى بالتطعيم، المضادات الحيوية، وتناول كميات كبيرة من السوائل، مكملات حمض الفوليك، وأدوية الألم.[5][9] قد تشمل التدابير الأخرى نقل الدم وأدوية هيدروكسي كارباميد.[5] يمكن علاج نسبة صغيرة من الناس عن طريق زرع خلايا نخاع العظام.[3]
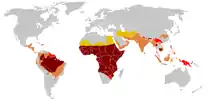
اعتبارًا من عام 2015، يعاني حوالي 4.4 مليون شخص من مرض الخلايا المنجلية، في حين يعاني 43 مليون شخص إضافي من سمة الخلايا المنجلية.[6][10] يعتقد أن حوالي 80٪ من حالات مرض الخلايا المنجلية تحدث في أفريقيا جنوب الصحراء الكبرى.[11] كما يحدث بشكل متكرر نسبيا في أجزاء من الهند، شبه الجزيرة العربية وبين الأشخاص من أصل أفريقي الذين يعيشون في أجزاء أخرى من العالم.[12] في عام 2015، أدى إلى حوالي 114800 حالة وفاة.[7] تم وصف الحالة لأول مرة في الأدبيات الطبية من قبل الطبيب الأمريكي جيمس ب. هيريك في عام 1910.[13][14] في عام 1949، تم تحديد انتقاله الجيني من قبل بيت، وفي. نيل.[14] في عام 1954، تم وصف التأثير الوقائي ضد الملاريا لسمة الخلايا المنجلية.[14]
أصل التسمية


وكلمة المنجلية مأخوذه من المنجل (الذي يحصد به النبات وفي بعض المناطق يطلق عليه المحش) وذلك لأن كريات الدم الحمراء تحت المجهر تأخذ شكل مقوس كالمنجل أو الهلال. وكلمة انيميا تعني فقر دم.
علم الأوبئة
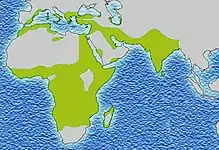
تم العثور على أعلى تردد من مرض فقر الدم المنجلي في المناطق المدارية، وخاصة أفريقيا جنوب الصحراء الكبرى والمناطق القبلية في الهند والشرق الأوسط.[15] هجرة أعداد غفيرة من هذه المناطق انتشار عالية للبلدان الانتشار المنخفض في أوروبا قد ازداد بشكل ملحوظ في العقود الأخيرة، وفي بعض الدول الأوروبية مرض الخلية المنجلية وقد تجاوز الآن حالات وراثية أكثر مألوفة مثل الناعور والتليف الكيسي.[16] وفي عام 2013 أدى إلى 176,000 حالة وفاة بسبب مرض الانيما المنجلي ارتفاعا من 113,000 حالة وفاة في عام 1990.[17]
يحدث مرض الخلية المنجلية أكثر شيوعا بين الأشخاص الذين يعيشون في المناطق الواقعة جنوب الصحراء الكبرى المدارية وشبه المدارية حيث الملاريا أو كان شائعا الأجداد. حيث الملاريا هو شائع، والتي تحمل أليل المنجلي واحد (سمة) يضفي ميزة انتقائية وبعبارة أخرى، لكونها متغايرة هو مفيد. على وجه التحديد، والبشر مع واحد من اثنين الأليلات من المعرض مرض الخلية المنجلية أعراض أقل حدة عند المصابين بالملاريا.[18]
أفريقيا
ثلاثة أرباع الحالات المنجلي تحدث في أفريقيا. وقدر تقرير منظمة الصحة العالمية مؤخرا أن حوالي 2٪ من الأطفال حديثي الولادة في نيجيريا تأثرت فقر الدم المنجلي، أي ما مجموعه 150,000 الأطفال المتضررين يولدون كل عام في نيجيريا وحدها. تردد الناقل يتراوح بين 10% و 40% في أفريقيا الاستوائية، وخفض إلى 1-2٪ على ساحل شمال افريقيا و<1٪ في جنوب أفريقيا.[19] وكانت هناك دراسات في أفريقيا أن تظهر انخفاضا كبيرا في معدل وفيات الرضع معدل تتراوح أعمارهم بين 2-16 شهرا، بسبب سمة المنجلي. حدث هذا في المناطق الغالبة من الحالات ملاريا.[20]
الولايات المتحدة الأمريكية
انتشار المرض في الولايات المتحدة ما يقرب من 1 في 5000، والتي تؤثر في الغالب الأمريكيين من أصل أفريقي جنوب الصحراء الكبرى، وفقا للمعهد الوطني للصحة.[21] وفي الولايات المتحدة، حوالي واحد من كل 500 الأميركيين الأفارقة الأطفال وواحد من بين كل 36000 طفل أسباني الأمريكية لديها فقر الدم المنجلي.[22] وتشير التقديرات إلى أن مرض فقر الدم المنجلي يؤثر 90000 الأميركيي.[23] ويتم تحديد معظم الأطفال مرض الانيما المنجلي الذين يعانون من الذين يولدوا في الولايات المتحدة الآن من قبل فحص الأطفال حديثي الولادة الروتينية. أربعة وأربعون ولاية إلى جنب مع مقاطعة كولومبيا وبورتوريكو وجزر فيرجن حاليا توفر فحص الأطفال حديثي الولادة ل مرض الانيما المنجلي[24][25] سمة الخلية المنجلية يحدث بين حوالي 1:12 الأمريكيين من أصل أفريقي و 1: 100 أسباني-امريكيين[26] وتشير التقديرات إلى أن 2.5 مليون أمريكي يحملون صفة الجين المنجلي المتخالف"التعاكس"[27]
فرنسا
نتيجة للنمو السكاني في مناطق أفريقيا والبحر الكاريبي فرنسا في الخارج والهجرة من شمال وأفريقيا جنوب الصحراء الكبرى إلى البر الرئيسى فرنسا، أصبح مرض الخلية المنجلية مشكلة صحية كبيرة في فرنسا.[28] مرض الانيما المنجلي أصبح الأكثر شيوعا الوراثية المرض في البلاد، مع انتشار الولادة العام لل1/2415 في البر الرئيسى فرنسا، في مقدمة"في الطليعة" بيلة الفينيل كيتون" مرض وراثي ويمثل بعدم القدرة على تحطيم-تكسير الفنيل الأنين الذي يسبب دمار وتلف في الدماغ والأعصاب إذا لم يعالج. (1/10862)، قصور الغدة الدرقية الخلقي (1/3132)، وتضخم الغدة الكظرية الخلقي (1/19008) والتليف الكيسي (1/5014) لنفس الفترة المشار إليها. في عام 2010، 31.5٪ من جميع الأطفال حديثي الولادة في البر الرئيسى"اليابسة" فرنسا (253466 من SCDأصل 805958) تم فحصهم ب (كانت هذه النسبة 19٪ في عام 2000). تم العثور على 341 حديثي الولادة معهم وناقلات متخالف 8744 تمثل 1.1٪ من جميع الأطفال حديثي الولادة في فرنسا. منطقة العاصمة باريس (إيل-دو-فرانس) هي المنطقة التي تمثل أكبر عدد من المواليد الجدد فحص ل مرض الانيما المنجلي "60٪ في عام 2010". على ثاني أكبر عدد من المعرضين للخطر في بروفانس ألب كوت دازور في ما يقرب من 43.2٪ وأقل عدد هو في بريتاني عند 5.5٪.[29][30]
المملكة المتحدة
في المملكة المتحدة ويعتقد أن ما بين 12,000 إلى 15,000 الناس لديهم مرض فقر الدم المنجلي[68] مع تقدير 250,000 حاملة للحالة في إنجلترا وحدها. كما يقدر عدد من شركات فقط، وجميع الأطفال حديثي الولادة في المملكة المتحدة تحصل على فحص الدم الروتيني للكشف عن هذه الحالة.[31] نظرا لكثير من البالغين في المجموعات المعرضة للخطر لا يعرفون إذا كانوا يحملون والنساء الحوامل والشركاء على حد سواء في وتقدم بضع الفرز حتى يتمكنوا من الحصول على المشورة إذا كان لديهم سمة فقر الدم المنجلي.[32] In addition blood donors from those in high risk groups are also screened to confirm whether they are carriers and whether their blood filters properly.[33] وبالإضافة إلى ذلك يتم فحص المتبرعين بالدم أيضا عن تلك الموجودة في المجموعات المعرضة للخطر لتأكيد ما إذا كانوا يحملون وعما إذا كانت المرشحات الدم "فلترة الدام" تعمل بشكل صحيح.[33] الجهات المانحة التي وجدت لتكون بعد ذلك يتم إبلاغ الناقلين ودمائهم، في حين غالبا ما تستخدم لأولئك نفس المجموعة العرقية، لا يتم استخدام لأولئك المصابين بمرض فقر الدم المنجلي الذين يحتاجون إلى نقل الدم.[34]
الشرق الأوسط
في المملكة العربية السعودية حوالي 4.2٪ من السكان تحمل سمة المنجلي و 0.26٪ يتلقى مرض الخلية المنجلية. أعلى نسبة انتشار في المنطقة الشرقية حيث تحمل ما يقرب من 17٪ من سكان الجينات و 1.2٪ لديهم مرض الخلية المنجلية.[35] في عام 2005 في المملكة العربية السعودية وبدأ اختبار قبل الزواج الإلزامي بما "HB electrophoresisفي ذلك اطلاق " حركة الجسيمات المشحونة في السائل أو الجل تحت تأثير المجال الكهربائي". وهو عبارة عن " ويهدف إلى تقليل الإصابة بمرض الانيما المنجلي والثلاسيميا.[36]
الهند ونيبال
مرض فقر الدم المنجلي هو شائع في الناس القبائل في وسط الهند الذين يشتركون في الربط الجينية مع السباق الأفريقي، حيث تراوح معدل انتشار 9,4 حتي 22,2٪ في المناطق الموبوءة ماديا براديش وراجستان وتشاتيسغار.[37] وهو أيضا منتشرة بين الناس ثارو من نيبال والهند، ولكن لديهم معدل الإصابة لسبع مرات أقل من الملاريا على الرغم من الذين يعيشون في منطقة تنتشر فيها الملاريا.[38]
جزر الكاريبي
في جامايكا، و 10٪ من السكان تحمل الجين المنجلي، مما يجعلها اضطراب وراثي الأكثر انتشارا في البلاد.[39]
أعراض المرض
وتكمن مشكلة المرض في إنتاج نخاع العظم لكريات دم حمراء - التي تنقل ثنائي الأكسجين O2 إلى مختلف أنحاء الجسم – غير طبيعية. نتيجة لخلل في تكوين الهيموجلوبين (خضاب الدم)وفي كميته أيضا.وهذه الخلايا غير الطبيعية تأخذ شكل المنجل وهي قابلة إلى التكسر وتتحلل بعد فترة قصيرة من إنتاجها وقد تعيق مرور الدم خلال الشعيرات الدموية، وقد تسد الأوعية الدموية فتسبب آلاما مبرحه في اجزاء مختلفة من الجسم خاصة في العظام منها عظام الاطرف والظهر.و قد تسد كريات الدم الحمراء المنجلية أي وعاء دموي في الرئتين أو في البطن أو حتى في المخ وقد تسبب مضاعفات خطيرة إضافة إلى الألام المبرحه التي يعاني منها الشخص المصاب. وأيضا الاضرار النفسية والاجتماعية للمريض وعدم القدرة على التحصيل العلمي والمعرفي بسبب تكرار دخوله للمستشفى بشكل شبة دائم. ويعتبر فقر الدم المنجلي من الأمراض الوراثية المزمنة. لما يسببة من آلام مبرحة وجدا قاسية. وعند حدوث نوبات الالم الشديدة لابد من استخدام العقاقير الطبية والمسكنات القوية.
قد يؤدي مرض الخلية المنجلية لحدوث مضاعفات الحادة والمزمنة المختلفة، والعديد منها لديها ارتفاع في معدل الوفيات.[40]
أزمة الخلايا المنجلية
المصطلح "أزمة الخلايا المنجلية" أو "أزمة التمنجل" يمكن استخدامها لوصف العديد من الحالات الحادة المستقلة التي تحدث في المرضى الذين يعانون من مرض الانيما المنجلي. نتائج مرض الانيما المنجلي في فقر الدم والأزمات التي قد تتكون من العديد من الأنواع بما في ذلك ازمة انسداد الأوعية الدموية، أزمة اللاتنسجية، أزمة تراكد الدم في الطحال، أزمة الدم الانحلالي، وغيرها. معظم نوائب أزمة التمنجل تستمر ما بين خمسة وسبعة أيام.[41] على الرغم من الإصابة، والجفاف، وحموضة الدم (وكلها لصالح عملية التمنجل) يمكن أن تكون بمثابة محفزات، في معظم الحالات، لا يوجد ظروف مهيئة لهذا المرض.[42]
أزمة انسداد الأوعية الدموية
سبب هذه الأزمة خلايا الدم الحمراء المنجلية التي تعرقل الشعيرات الدموية وتحد من تدفق الدم إلى العضو مما يؤدي إلى نقص الأكسجين، والألم، وموت الخلايا، وغالبا ما تلف العضو.التكرار والشدة والمدة لهذه الأزمات تختلف إلى حد كبير. يتم التعامل مع الأزمات المؤلمة مع الإكثار من الماء، والمسكنات، ونقل الدم. يتطلب إدارة الألم تناول الأفيونات على فترات منتظمة حتى انقشاع الأزمة. لأزمات أكثر اعتدالا، مجموعة فرعية من المرضى تعمل على إدارة الألم باستخدام الأدوية غير الستيرويدية المضادة للالتهاب (مثل ديكلوفيناك أو نابروكسين). لأزمات أشد، فإن معظم المرضى يحتاجون إلى إدارة الألم بأخذ الافيونات في الوريد. وتستخدم أجهزة تسكين التي تسيطر على المريض عادة في هذا الإعداد. أزمة انسداد الأوعية الدموية التي تنطوي على أجهزة مثل القضيب[43] أو تعتبر الرئتين حالة الطوارئ وتعالج مع نقل خلايا الدم الحمراء. آلة تحسين التنفس، هي تقنية موصى بها لتشجيع التنفس العميق للحد من تطوير انهيار وإغلاق الرئة.[44]
أزمة تراكد الدم في الطحال
بسبب أوعيتها الضيقة ووظيفتها في تطهير خلايا الدم الحمراء التالفة، كثيرا ما تتأثر الطحال.[45] وعادة ما تموت أنسجتها قبل نهاية مرحلة الطفولة في الأفراد الذين يعانون من فقر الدم المنجلي.هذا الضرر الطحال يزيد من خطر العدوى من الكائنات البكتيرية المحاطة ب كبسولة للحماية.[46][47] ينصح المضادات الحيوية الوقائية والتطعيمات لأولئك الذين يفتقرون إلى وظيفة الطحال المناسبة.
ازمة تراكد الدم في الطحال هي توسعات مؤلمة حادة في الطحال، والناجمة عن محاصرة خلايا الدم الحمراء داخل الطحال مما أدى إلى انخفاض حاد في مستويات الهيموجلوبين مع احتمال نقص حاد في حجم الدم. تعتبر هذه الأزمة حالة طارئة. إذا لم يعالج، المرضى قد يموتون في غضون 1-2 ساعات بسبب فشل في الدورة الدموية. احيانا يمكن إدارة الوضع بنقل الدم. هذه الأزمات هي عابرة، فإنها تستمر لمدة 3-4 ساعات، ويمكن أن تستمر ليوم واحد.[48]
أزمة الصدر الحادة
يتم تعريف متلازمة الصدر الحادة من قبل اثنين على الأقل من العلامات التالية أو الأعراض: ألم في الصدر، الحمى، ارتشاح رئوي أو شذوذ بؤري، أعراض تنفسية، أو نقص الأكسجين.[49] ومن المضاعفات الثانية الأكثر شيوعا وحساب نحو 25٪ من الوفيات في المرضى الذين يعانون من مرض الانيميا المنجلية، معظم الحالات الحالية مع أزمة انسداد الأوعية الدموية ثم يتطورون لحدوث أزمة الصدر الحاد[50][51] ، مع ذلك’ حوالي 80٪ من المرضى الذين لديهم أزمة انسداد الأوعية الدموية أثناء وجود أزمة الصدر الحاد.
أزمة اللاتنسجية
أزمة اللاتنسجية هي تدهورات حادة من فقر الدم، وتنتج مظهر شاحب، وسرعة في دقات القلب، والتعب. يتم تشغيل هذه الأزمة عادة باستخدام ب19، والتي تؤثر بشكل مباشر على إنتاج خلايا الدم الحمراء عن طريق غزو سوالف الخلايا الحمراء ومضاعفاتها وتدميرها.[52] العدوى الفيروسية يمنع تماما تقريبا إنتاج خلايا الدم الحمراء لمدة يومين أو ثلاثة أيام. في الأفراد الأسوياء، وهذا هو نتيجة تذكر، ولكن نتائج قصر عمر الخلية الحمراء من مرضى الانيميا المنجلية هي حالة مفاجئة تهدد الحياة . انخفاض تعداد الخلايا الشبكية (الخلايا الحمراء غير الناضجة) بشكل كبير خلال المرض (يسبب نقصان غير طبيعي في خلايا الدم غير الناضجة)، وسرعة دوران خلايا الدم الحمراء يؤدي إلى انخفاض في الهيموغلوبين. هذه الأزمة يستغرق 4 أيام إلى أسبوع واحد لتختفي. معظم المرضى يمكن أن تدار داعمة. بعضهم بحاجة لنقل الدم.[53]
أزمة الدم الانحلالي
الأزمات الدم الانحلالي هي نزول متسارع حاد في مستوى الهيموغلوبين. حيث تكسر خلايا الدم الحمراء بمعدل أسرع. وهذا أمر شائع خاصة في المرضى الذين يعانون من نقص G6PD في الوجود (مرض التفول).[54] احيانا عملية إدارته عملية داعمة، وذلك مع عمليات نقل الدم.[44]
أخرى
واحدة من أقدم المظاهر السريرية هو التهاب الأصابع، كما عرض في وقت مبكر من ستة أشهر من العمر، ويمكن أن يحدث في الأطفال الذين يعانون من سمة المنجلية.[55] إن الأزمة يمكن أن تصل إلى شهر واحد.[56] وثمة نوع آخر معترف به من أزمة المنجلية ومتلازمة الصدر الحادة، ويتميز بحمى وألم في الصدر، صعوبة في التنفس، والارتشاحات الرئوية على الصدر بالأشعة السينية. وبالنظر إلى أن الالتهاب الرئوي والتمنجل في الرئة على حد سواء يمكن إنتاج هذه الأعراض، يتم التعامل مع المريض على حد سواء الظروف.[57] ويمكن أن يكون سببها أزمة مؤلمة، عدوى الجهاز التنفسي، انصمام نخاع العظم، أو ربما عن طريق الانخماص، إدارة المواد الأفيونية، أو الجراحة.
علم الوراثة

عادة، البشر لديهم الهيموجلوبين A، التي تتألف من اثنين ألفا وسلسلتي بيتا، الهيموغلوبين A2، والتي تتألف من اثنين ألفا واثنين من سلاسل الدلتا، والهيموجلوبين F، تتكون من اثنين ألفا وسلسلتين جاما في أجسامهم. من هذه، الهيموجلوبين F تهيمن حتى حوالي 6 أسابيع من العمر ثم A تهيمن في جميع مراحل الحياة.
شروط المنجلي لديها نمط مقهورة من الميراث من الآباء والأمهات. أنواع الهيموجلوبين شخص يجعل في خلايا الدم الحمراء تعتمد على جينات الهيموغلوبين الموروثة من والديه. إذا كان أحد الأبوين فقر الدم المنجلي والآخر لديه سمة المنجلي، ثم الطفل لديه فرصة 50٪ من وجود مرض الخلايا المنجلية وفرصة 50٪ من وجود سمة المنجلي. عندما يكون كلا الوالدين سمة المنجلي، وهو طفل لديه فرصة 25٪ من أمراض الدم المنجلي، و 25٪ لا تحمل أي أليل المنجلي، و 50٪ لديهم حالة متخالف.
طفرة جين اخلية المنجلية ربما نشأت عفويا في مناطق جغرافية مختلفة، كما اقترح تحليل اقتطاع نوكلياز داخلية. وتعرف هذه المتغيرات كما الكاميرون والسنغال وبنين والبانتو، والمملكة العربية الآسيوية. هذه الأهمية السريرية لأن ترتبط بعض مع أعلى مستويات هيموغلوبين f، على سبيل المثال، والسنغال، والمتغيرات السعودية الآسيوية، وتميل إلى أن تكون أكثر اعتدالا في المرض.[58]
في الناس متخالف hgbs(ناقلات من الهيموجلوبين المتمنجل)، مشاكل البلمرة طفيفة، وذلك لأن أليل (allele) "واحد من اثنين أو أكثر من الأشكال البديلة من الجين والذي ينشأ عن طريق الطفرات وتوجد في نفس المكان على كروموسوم" العادي قادر على إنتاج أكثر من 50٪ من الهيموجلوبين. في الناس متماثل لHgbS، وجود سلسلة طويلة من البوليميرات من HBS تشوه شكل خلايا الدم الحمراء من سلسة مثل شكل دونات لخشنة وكامل"مليء" من المسامير، مما يجعلها هشة وعرضة للكسر داخل الشعيرات الدموية. ناقلات لديهم أعراض إلا إذا يحرمون من الأكسجين (على سبيل المثال، في حين تسلق الجبال) أو أثناء الجفاف الشديد. يحدث مرض الخلية المنجلية عندما يتم استبدال الحمض الأميني السادس، وحمض الجلوتاميك، من خلال فالين لتغيير بنيتها ووظيفتها. على هذا النحو، وكما هو معروف فقر الدم المنجلي كما E6V. فالينحمض اميني هو مسعور، مما تسبب في الهيموجلوبين للانهيار على نفسه في بعض الأحيان. لم يتم تغيير بنية خلاف ذلك. عندما ينهار كمية كافية من الهيموغلوبين على نفسها خلايا الدم الحمراء تصبح منجلية الشكل.
الخلل الجيني هو طفرة معروفة لنيوكليوتيد منفرد (انظر إحدى النوكليوتيدات من السلسلة) (A to T) من جين غلوبين β، والذي ينتج حمض الجلوتاميك يجري الاستعاضة عن فالين في الموقع رقم 6. لاحظ، وضع الترقيم التاريخي هذه بقايا حمض الجلوتاميك في موقف 6 بسبب تخطي ميثيونين الكودون الأول في موقع الترقيم لحمض الاميني للبروتين. وتدعو التسميات الحالية لحساب ميثيونين كأول الأحماض الأمينية، مما أدى إلى بقايا حمض الجلوتاميك الوقوع في موقف 7. العديد من المراجع لا تزال تشير إلى موقف 6 وكلا أن من المحتمل أن يكون مرجعا للوضوح. يشار الهيموغلوبين S مع هذه الطفرة على أنها HBS، على عكس الكبار العادي HBA. ويرجع ذلك إلى طفرة من النوكليوتيدات واحدة من اضطراب وراثي، من GAG لGTG كودون على حبلا الترميز، والذي كتب من حبلا القالب إلى كودون GUG. هذا هو عادة طفرة حميدة، مما تسبب في عدم وجود آثار واضحة على الهياكل الثانوية، الثلاثية، أو الرباعية للهيموغلوبين في ظروف تركيز الأكسجين العادي. الذي تسمح في ظل ظروف من تركيز الأكسجين منخفضة، هو بلمرة هيموغلوبين (HbS) S نفسها. الشكل غير المؤكسج من الهيموجلوبين يعرض على رقعه مسعورة على البروتين بين لوالب E و F . سلسلة الجانب الدهني"كربوني" الذي لا يذوب مع الماء "التي تميل إلى صد أو فشل الخلط مع المياه." من بقايا حمض أميني أساسي في موقف 6 من سلسلة بيتا في الهيموجلوبين قادرة على ربط مع الرقعة المسعورة*"الدهني-الكربوني الذي لا يختلط ولا يذوب مع الماء"، مما تسبب في جزيئات الهيموغلوبين S لتتجمع وتشكيل رواسب ليفية.
-الأليل "واحد من اثنين أو أكثر من أشكال بديلة من الجين الذي ينشأ عن طريق الطفرات وتوجد في نفس المكان على كروموسوم." المسؤولة عن فقر الدم المنجلي يمكن العثور عليه في الذراع القصير للكروموسوم 11، وبشكل أكثر تحديدا 11p15. والشخص الذي يحصل على الجينات المعيبة من كل من الأب والأم تطور المرض. الشخص الذي يحصل اليل واحد معيب وواحد أليل صحي لا يزال بصحة جيدة، ولكن يمكن أن تمر على هذا المرض وكما هو معروف بالناقل أو متغايرة الزيجوت. متغايرة الزيجوت لا تزال قادرة على التعاقد الملاريا، ولكن الأعراض عادة ما تكون أقل حدة.[59]
يرجع ذلك إلى ميزة التكيف في متغايرة، وهذا المرض لا يزال سائدا، خاصة بين الأشخاص الذين يعانون من أصل مؤخرا في المناطق المنكوبة الملاريا، مثل أفريقيا والبحر الأبيض المتوسط والهند والشرق الأوسط.[60] وكان تتوطن فيها الملاريا تاريخيا للجنوب أوروبا، ولكن تم الإعلان عن القضاء عليها في منتصف القرن 20، باستثناء حالات متفرقة نادرة.[61]
طفيلي الملاريا لديه دورة حياة معقدة وينفق جزء منها في خلايا الدم الحمراء. في الناقل، وجود طفيل الملاريا يتسبب في خلايا الدم الحمراء مع الهيموغلوبين معيب للتمزق قبل الأوان، مما يجعل من الطفيليات المتصورة (بلازموديوم) غير قادرة على التكاثر. وعلاوة على ذلك، بلمرة الهيموغلوبين يؤثر على قدرة الطفيل على هضم الهيموغلوبين في المقام الأول. لذلك، في المناطق التي الملاريا فيها مشكلة، فرص الناس من البقاء على قيد الحياة تزيد في الواقع إذا كانت تحمل سمة المنجلي (الاختيار-الانتقاء للمتغايرة المختلفة).
في الولايات المتحدة الأمريكية، مع عدم وجود مرض الملاريا، وانتشار فقر الدم المنجلي بين السود أقل (حوالي 0.25٪) منها في غرب أفريقيا (نحو 4.0٪)، مع النزول المستمر. دون وجود الملاريا، طفرة الخلايا المنجلية هي ضارة، وتميل إلى الانخفاض في السكان المتضررين من الانتقاء الطبيعي، والآن بشكل مصطنع من خلال الفحص الجيني ما قبل الولادة. ومع ذلك، ينحدر المجتمع الأمريكيين من أصل أفريقي من خليط كبير من عدة مجموعات عرقية أفريقية وغير أفريقية، ويمثل أيضا أحفاد الناجين من الاسترقاق وتجارة الرقيق. وهكذا، وعلى درجة أقل من زواج الأقارب، وخاصة، وارتفاع ضغط غير طبيعي الصحي انتقائية من خلال العبودية قد تكون التفسيرات الأكثر قبولا لانخفاض معدل انتشار فقر الدم المنجلي (وربما غيرها من الأمراض الوراثية) بين الأمريكيين من أصل أفريقي مقارنة شبه الأفارقة جنوب الصحراء الكبرى. وثمة عامل آخر يحد من انتشار جينات الدم المنجلي في أمريكا الشمالية هو عدم وجود الميول الثقافية لتعدد الزوجات، والذي يسمح للذكور المتضررة على مواصلة البحث عن الأطفال تتأثر مع عدة شركاء.[62]
الفسيولوجيا المرضية

فقدان مرونة خلايا الدم الحمراء هي المركزية إلى الفيسيولوجيا المرضية لمرض الخلية المنجلية. خلايا الدم الحمراء العادية هي مرنة جدا، والذي يسمح للخلايا لتغيير شكلها للمرور عبر الشعيرات الدموية. في مرض الخلية المنجلية، انخفاض تركيز الاكسجين يعزز تمنجل خلايا الدم الحمراء ونوبات متكررة من التمنجل تؤدي إلى تلف غشاء الخلية وانخفاض مرونة الخلية. تفشل هذه الخلايا للعودة إلى الشكل الطبيعي عند استعادة التركيز الطبيعي للأكسجين. ونتيجة لذلك، وهذه الخلايا في الدم جامدة غير قادرة على تغيير شكلها لأنها تمر من خلال الشعيرات الدموية الضيقة، مما يؤدي إلى انسداد الأوعية ونقص الأوكسجين.
ويتسبب فقر الدم الفعلي للمرض من قبل انحلال الدم، وتدمير خلايا الدم الحمراء، بسبب شكلها. على الرغم من أن نخاع العظم يحاول تعويض من خلال خلق خلايا الدم الحمراء جديدة، فإنه لا تتناسب مع معدل الدمار.[63] خلايا الدم الحمراء الصحية تعمل عادة ل90-120 يوما، ولكن الخلايا المنجلية فقط 10-20 أيام.[64]
التشخيص
في هيموغلوبين المتوافق، وتعداد الدم الكامل يكشف مستويات الهيموجلوبين في حدود 6-8 غ / ديسي لتر مع عدد الخلايا الشبكية عالية (كما يعوض نخاع العظام لتدمير الخلايا المنجلية من خلال إنتاج المزيد من خلايا الدم الحمراء). في أشكال أخرى من أمراض الدم المنجلي، ومستويات الهيموغلوبين تميل إلى أن تكون أعلى من ذلك. فيلم الدم قد تظهر ملامح قصور الطحال (الخلايا المستهدفة والاجسام هاول-جولي).
تمنجل خلايا الدم الحمراء على فيلم الدم، يمكن أن يتسبب من خلال إضافة ميتابيسلفيت الصوديوم. ويمكن أيضا وجود الهيموجلوبين المنجلي ان يظهر مع "اختبار الذوبان المنجلية". مزيج من الهيموغلوبين س (الهيموغلوبين س) في محلول الحد (مثل دايثيونات الصوديوم) يعطي مظهر العكرة، في حين الهيموغلوبين الطبيعي يعطي محلولا واضحا.
ويمكن الكشف عن أشكال الهيموجلوبين غير طبيعية على الهيموجلوبين الكهربائي، وهو شكل من هلام الكهربائي Cالذي مختلف أنواع الهيموجلوبين تتحرك بسرعات متفاوتة. الهيموجلوبين المنجلي والهيموجلوبين سي مع التمنجل (HgbSC) وهما الأكثر شيوعا أشكال يمكن التعرف عليها من هناك. يمكن تأكيد التشخيص والتفريق اللوني السائل عالي الاداء. نادرا ما يتم تنفيذ الاختبارات الجينية، من تحقيقات أخرى هي محددة للغاية للهيموغلوبين (س وسي) HbS and HbC.[65]
وغالبا ما عجلت أزمة الخلايا المنجلية الحادة عن طريق العدوى. ولذلك، فإن تحليل البول للكشف عن وجود عدوى المسالك البولية غامض، والصدر بالأشعة السينية للبحث عن الالتهاب الرئوي غامض، ينبغي أن يقام به بشكل روتيني.[66]
الناس الذين يعرفون بحاملة للمرض غالبا ما يخضع الاستشارة الوراثية قبل أن إنجاب طفل. اختبار لمعرفة ما إذا كان الجنين قد يأخذ المرض إما عينة دم من الجنين أو عينة من السائل الأمنيوسي. منذ أخذ عينة دم من الجنين لديها مخاطر أكبر، وعادة ما يستخدم الاختبار الأخير. يوفر فحص الأطفال حديثي الولادة ليس فقط وسيلة للكشف المبكر للأفراد المصابين بأمراض الدم المنجلي، ولكن يسمح أيضا للتعرف على مجموعات من الناس التي تحمل سمة فقر الدم المنجلي.[67]
المعالجة
حمض الفوليك والبنسلين
الأطفال الذين ولدوا مع مرض الخلية المنجلية تخضع للمراقبة من طبيب الأطفال وتتطلب معالجة من قبل أمراض الدم للتأكد من أنها لا تزال في صحة جيدة. هؤلاء المرضى تأخذ جرعة 1 ملغ من حمض الفوليك يوميا لمدى الحياة. من الولادة وحتى سن الخامسة، كما أن لها أن تأخذ البنسلين يوميا بسبب نظام المناعة غير الناضجة الذي يجعلها أكثر عرضة لأمراض الطفولة المبكرة.
الوقاية الكيميائية الملاريا
لا ينطبق تأثير وقائي من سمة المنجلي للأشخاص الذين يعانون مرض الخلية المنجلية. في الواقع، هم أكثر عرضة للإصابة بالملاريا، لأن السبب الأكثر شيوعا من الأزمات المؤلمة في بلدان الملاريا هو الإصابة بالملاريا. ولذلك فمن المستحسن أن الأشخاص الذين يعانون من مرض الخلية المنجلية الذين يعيشون في بلدان الملاريا يجب أن يتلقى الوقاية الكيميائية المضادة للملاريا للحياة.[68]
ازمة انسداد الاوعية الدموية
معظم الناس يعانون من مرض فقر الدم المنجلي والحلقات مؤلمة بشكل مكثف دعت بأزمة انسداد الاوعية الدموية. ومع ذلك، فإن تكرار وشدة ومدة هذه الأزمات تختلف بشكل كبير. يتم التعامل مع الأزمات المؤلمة عرضيا مع أدوية الألم. يتطلب معالجة الألم معالجة أفيونية على فترات منتظمة حتى انقشاع الأزمة. لأزمات أكثر اعتدالا، مجموعة فرعية من المرضى على إدارة المسكنات (مثل ديكلوفيناك أو نابروكسين). لأزمات أشد، فإن (PCA)معظم المرضى يحتاجون إلى معالجة للمرضى بادخال أفيونات في الوريد. وتستخدم تسكين الأجهزة التي تسيطر عليها المريض عادة في هذا الإعداد. دايفينهايدرامين هو أيضا عامل فعال أن الأطباء كثيرا ما يصف للمساعدة في التحكم في الحكة المرتبطة باستخدام المواد الأفيونية.
أزمة حادة في الصدر
معالجة مشابهة لأزمة انسداد الاوعية الدموية، مع إضافة المضادات الحيوية (عادة الكينولون أو ماكرولايد، منذ جدار الخلية التي تعاني من نقص ويعتقد ["شاذة"] البكتيريا للمساهمة في متلازمة)[69] مكملات الأكسجين لنقص الأكسجين، ومراقبة قريبة. يجب أن تتفاقم الارتشاح الرئوي أو زيادة متطلبات الأكسجين، بحيث يشار إلى نقل دم بسيط أو تبديل الدم. ينطوي هذا الأخير تبادل لجزء كبير من الكتلة المرضية من الخلايا الحمراء العادية، Sمما يقلل من نسبة الهيموغلوبين المنجلي في دم المريض. المريض الذي يشتبه اصابته بمتلازمة الصدر الحادة يجب أن يتم قبولهم" ابقاءهم "بالمستشفى مع خطر تفاقم الموشر "التدريج أ" الذي يشير إلى إدخالهم وحدة العناية المركزية.[49]
هيدروكسي يوريا
أول دواء تمت الموافقة عليه لعلاج فقر الدم المنجلي ، هيدروكسي يوريا، وقد تبين خفض عدد وشدة الهجمات [70] Charache et al في دراسة أجريت في 1995 بواسطة وتبين أن من المحتمل زيادة وقت بقاء في دراسة في عام2003 شتاينبرغ وآخرون.[71] ويتحقق ذلك، جزئيا، عن طريق إعادة تنشيط إنتاج الهيموغلوبين الجنيني بدلا من الهيموغلوبين (س) يسبب فقر الدم المنجلي. هيدروكسي يوريا سبق استخدامها كعامل العلاج الكيميائي، وهناك بعض القلق من أن الاستخدام طويل الأمد قد تكون ضارة، ولكن ثبت هذا الخطر أن يكون إما غائبة أو صغيرة جدا وأنه من المرجح أن الفوائد تفوق المخاطر.[40][72]
العلاج بنقل الدم
وغالبا ما تستخدم عمليات نقل الدم في إدارة مرض الخلايا المنجلية في الحالات الحادة ومنع حدوث مضاعفات من خلال خفض عدد خلايا الدم الحمراء التي يمكن أن المنجل بإضافة خلايا الدم الحمراء العادية.[73] وفي الأطفال وقائية الدم الحمراء المزمن وقد تبين أن تكون فعالة إلى حد ما في الحد من خطر السكتة الدماغية الأولى أو السكتة الدماغية الصامتة عندما يظهر دوبلر عبر الجمجمة الموجات فوق الصوتية غير طبيعية زيادة الدماغية سرعات تدفق الدم. في أولئك الذين أصيبوا حدث السكتة الدماغية مسبق كما أنه يقلل من خطر الإصابة بالسكتة الدماغية والسكتات الدماغية الصامتة المتكررة إضافية.[74][75]
زرع نخاع العظم
وقد أثبتت عمليات زرع نخاع العظم فعال في الأطفال. زرع نخاع العظم هي العلاج الوحيد المعروف له. ومع ذلك، زرع نخاع العظم.[76] HLA مع ذلك، زرع نخاع العظم من الصعب الحصول على لنوع/صنف المحدد اللازم. من الناحية المثالية، فإن أعضاء الأسرة التوأم (مسانج/ماثلة جينيا أو متطابقة، وبالتالي متوافقة مناعيا، وخاصة المتعلقة بشكل وثيق أن زرع لا تثير استجابة مناعية.) أو قريب (خيفي/الممنوحة أو مترع بها أي التي تنطوي على الأنسجة أو الخلايا التي تختلف وراثيا، وبالتالي تتعارض مناعيا، على الرغم من الأفراد من نفس النوع.) تبرع نخاع العظام الضروري للزرع.[77]
الطقس
حوالي 90٪ من المرضى البقاء على قيد الحياة إلى سن 20، وعلى مقربة من 50٪ البقاء على قيد الحياة بعد انقضاء العقد الخامس.[78] وفي عام 2001، وفقا لإحدى الدراسات التي أجريت في جامايكا، وكان يعني البقاء على قيد الحياة المقدرة لمرضى فقر الدم المنجلي 53 سنة للرجال و 58 سنة للنساء مع متماثل.
مضاعفات
فقر الدم المنجلي يمكن أن يؤدي إلى مضاعفات مختلفة، بما في ذلك:
زيادة خطر العدوى البكتيرية شديدة بسبب فقدان الأنسجة يعمل الطحال (وقابلة للمقارنة لخطر العدوى بعد أن الطحال جراحيا). عادة ما تحدث هذه العدوى عن طريق الكائنات مغلفة مثل العقدية الرئوية والمستدمية النزلية. الوقاية اليومية بالبنسلين هو العلاج الأكثر شيوعا في مرحلة الطفولة، مع بعض مواصلة العلاج إلى أجل غير مسمى. المرضى الذين يستفيدون اليوم من التطعيم الروتيني لل.(س) الرئوية.[79]
السكتة الدماغية، والتي يمكن أن تنجم عن التآكل التدريجي للأوعية الدموية، ويمنع الاكسجين من الوصول إلى المخ. يحدث احتشاء الدماغ لدى الأطفال والنزيف الدماغي في البالغين. [بحاجة لمصدر]
السكتة الدماغية الصامتة تسبب أي أعراض فورية، ولكن يرتبط مع تلف في خلايا المخ. السكتة الدماغية الصامتة وربما خمسة أضعاف المشتركة كما السكتة الدماغية الأعراض. حوالي 10-15٪ من الأطفال مرض الانيما المنجلي والذين يعانون من يعانون السكتات الدماغية، مع السكتات الدماغية الصامتة السائدة في المرضى الأصغر سنا.[80][81]
تحص صفراوي (حصى في المرارة) والتهاب المرارة قد تنجم عن إنتاج البيليروبين الزائد بسبب انحلال الدم لفترة طويلة. نخر اوعائي (نخر العظام العقيم) من الورك والمفاصل الرئيسية الأخرى قد تحدث نتيجة لنقص الأوكسجين.[82] انخفضت التفاعلات المناعية نتيجة لقصور الطحال (خلل في الطحال) [83] فحولة واحتشاء القضيب [84] التهاب العظم والنقي (عدوى بكتيرية العظام)، والسبب الأكثر شيوعا لالتهاب العظم والنقي في مرض الانيما المنجلي هو السالمونيلا (وخاصة الأنماط المصلية شاذة السالمونيلا التيفية الفأرية، السالمونيلا الملهبة، كوليرا السالمونيلا، السالمونيلا التيفية ب)، تليها المكورات العنقودية الذهبية والعصيات المعوية سلبية الغرام ربما بسبب التمنجل داخل الأوعية الدموية من الأمعاء يؤدي إلى احتشاء دماغي غير مكتمل.[85]
يمكن أن يحدث التسامح المواد الأفيونية كما طبيعية، والاستجابة الفسيولوجية للاستخدام العلاجي للمواد الأفيونية. الإدمان على المواد المخدرة لم يحدث أكثر شيوعا بين الأفراد يعانون من مرض فقر الدم المنجلي من بين غيرهم من الأفراد تعامل مع المواد الأفيونية لأسباب أخرى. [بحاجة لمصدر]
- نخر حليمي حاد في الكلى
- قرحة الساق[86]
- في العيون، واعتلال الشبكية الخلفية، واعتلال الشبكية التكاثري، نزف الزجاجي، وانفصال الشبكية يمكن أن يؤدي إلى العمى.[87] ينصح بفحص منتظم للعين سنوياً
خلال فترة الحمل، تأخر النمو داخل الرحم، والإجهاض العفوي، وتسمم الحمل الألم المزمن: وحتى في حالة عدم وجود الألم الحاد فازو-مسد، وكثير من المرضى الذين لديهم ألم مزمن غير المبلغ عنه
ارتفاع ضغط الدم الرئوي (زيادة الضغط على الشريان الرئوي) يمكن أن يؤدي إلى الضغط على البطين الأيمن ومخاطر فشل القلب؛ الأعراض النموذجية هي ضيق في التنفس، وانخفاض القدرة على التحمل، وحلقات إغماء [بحاجة لمصدر] 21٪ من الأطفال و 30٪ من البالغين لديهم دليل على ارتفاع ضغط الدم الرئوي عند اختباره"عند الفحص"؛ هذا ويرتبط مع انخفاض مسافة قريبة وزيادة معدل الوفيات.[88]
الفشل الكلوي المزمن بسبب اعتلال الكلية المنجلي يتجلى مع ارتفاع ضغط الدم، وفقدان البروتين في البول، وفقدان خلايا الدم الحمراء في البول وتفاقم فقر الدم. إذا كان يتقدم حتى نهاية مرحلة الفشل الكلوي، فإنه يحمل بسوء التشخيص.[89]
سبب المرض
سبب حدوث هذا المرض هو حدوث خلل وراثي أثناء اصطناع الهيموغلوبين (خضاب الدم) في الجسم. يتألف الهيموغلوبين بروتين كرية الدم الحمراء من زوجين من سلاسل البولي ببتايد تتألف من 141 حمض أميني على سلسلة و146 على الأخرى، مما يشكل بالإجمال 574 وحدة حمض أميني في السلاسل الأربعة والتي لكل منها موقع محدد. يحدث المرض عند اصطناع الهيموغلوبين والذي يحصل على حمض نووي ريبوزي رسول حيث يحل GUG مكان GAG بالتالي يحل الفالين محل حمض الغلوتاميك في مكانه الخاص، بالتالي فإن الروابط الهيدروجينية التي تشكل البنى الأعلى من الهيموغلوبين تعطي شكل غير طبيعي للبروتين المتشكل. hba هموغلوبين طبيعي hbs هموغلوبين غير طبيعي إلى ما تعود A و B?
ويعتبر جهاز (VARIANT) من صناعة شركة بيوراد من أهم الأجهزة المرجعية في العالم لفحص هذا المرض .
مضاعفات المرض
- الدوار والدوخة عند القيام
- الغشيان عند السقوط من مكان شبه عالي
- متلازمة الصدر الحادة
- قصور القلب
- قصور كلوي مزمن
- ارتفاع ضغط الشريان الرئوي
- تكوين حصوات مرارية
- تضخم وقصور في الطحال
- ضعف المناعة
- تلف مع خشونة في مفصل الورك
- جلطة الدماغ
- تقرحات قدمية مزمنة
- اعتلال شبكية العين
- الوفاة
علاج المرض
تختلف شدة المرض من شخص إلى اخر. غالبا ما ينقص المرض من عمر المريض الافتراضي إلا أن هناك أناس معمرين بهذا المرض.
- على المريض تناول الأطعمة التي تحتوي على نسبة أكبر من الحديد مثل الأكباد بأنواعها ككبدة الإبل والبقر والغنم حتى كبدة الدجاج وكذلك الكلاوي والطحال والقلب، وتجد من الأطباء الحاذقين من يصف هذه الأشياء للحوامل لتقوية العظام عند الجنين وكذلك تناول الأسماك وصفار البيض.
- تناول الخضروات والفواكه التي تحتوي على الحديد على وجه الخصوص مثل السبانخ والبقدونس وكذلك العدس والفاصوليا.
- شرب الكثير من السوائل مثل شراب البنجر وأخص شرب فيتامين حمض الفوليك يوميا ولو تناولت المريض أو المريضة كبسولة حمض الفوليك 5ملجم لكان هذا نافع جداً أما بالنسبة لشرب الحليب فقد ساد الاعتقاد أنه مصدر للحديد وهذا غير صحيح، نعم فيه نسبة من الحديد ولكن لايعد مصدر أساسي ولكن فيه عنصر الكالسيوم الذي يساعد على امتصاص الحديد ومثله الأجبان والألبان.
- يصرف دواء الهيدروكسي يوريا إذا كان المرض شديد. الشفاء من المرض لا يمكن الا بزراعة نخاع عظم جديد وهذا الخيار متاح لفئة قليلة من المرضى وخصوصا الأطفال.
التاريخ
التقرير الحديث الأول من مرض الخلية المنجلية قد تكون في عام 1846، حيث تمت مناقشة عملية التشريح من العبد هارب تنفيذها. وكانت النتائج الرئيسية غياب الطحال[90][91] وكانت هناك أيضا تقارير بين العبيد الأفارقة في الولايات المتحدة واظهار مقاومة للملاريا ولكن يجري عرضة للقرحات الساق.[91] الخصائص غير طبيعية من خلايا الدم الحمراء، التي قدمت في وقت لاحق اسمهم إلى الشرط، وصفت لأول مرة من قبل إرنست إدوارد الحديد (1877-1959)، المتدرب إلى القلب شيكاغو وأستاذ الطب جيمس ب. هيريك (1861-1954)، في عام 1910. شوهد الحديد"غريب ممدود و "الخلايا على شكل المنجل في دم رجل يدعى والتر كليمنت نويل، في السنة الأولى وهو طالب يبلغ من العمر 20 عاما الأسنان من غرينادا. ونويل تم قبولهم في مستشفى شيكاغو المشيخية في ديسمبر 1904 يعانون من فقر الدم.[92][93] وقد أعيدوا نويل عدة مرات على مدى السنوات الثلاث المقبلة ل "الروماتيزم العضلي" و "هجمات صفراوي" ولكن أكمل دراسته وعاد إلى عاصمة غرينادا (سانت جورج) لممارسة طب الأسنان. مات من الالتهاب الرئوي في عام 1916 ودفنت في مقبرة كاثوليكية في في شمال غرينادا.[93][94] بعد فترة وجيزة تقرير هيريك، ظهرت قضية أخرى في ولاية فرجينيا الطبية نصف شهرية مع نفس العنوان " غريب مطول والمنجل على شكل كريات الدم الحمراء في حالة فقر شديد ". ويستند هذا المقال على المريض ادخل إلى مستشفى جامعة فرجينيا في 15 نوفمبر، 1910.[95] وفي وصف لاحق من قبل فيرن ميسون في عام 1922، اسم "فقر الدم المنجلي" واستخدم لأول مرة.[96][97] مشاكل الطفولة تتصل الخلايا المنجلية لم يتم الإبلاغ عنها المرض حتى 1930 على الرغم من أن هذا لا يمكن أن يكون من غير المألوف في السكان من أصل إفريقي.[91]
ممفيس الطبيب يمل تايلور، وهو باحث غزير في مرض فقر الدم المنجلي، لأول مرة التمييز بين مرض فقر الدم المنجلي وسمة في عام 1933، على الرغم من أن الأمر استغرق حتى 1949 حتى تم توضيح الخصائص الجينية بواسطة جيمس ف نيل وبنجر 1949 كان العام عندما وصف لينوس بولينغ في السلوك الكيميائي غير عادي من الهيموغلوبين" س"وعزا ذلك إلى خلل في الجزيء نفسه.[98] وقد وصفت التغيير الجزيئي الفعلي في هيموجلوبين س في أواخر عام 1950 بواسطة فيرنون انجرام وقال انه في وقت متأخر 1940 وأوائل عام 1950 شهدت مزيدا من التفاهم في العلاقة بين الملاريا ومرض فقر الدم المنجلي. في عام 1954، وإدخال الهيموجلوبين الكهربائي يسمح اكتشاف فرعية معينة، مثل مرض HbSC مرض الانيميا المنجلية.
وأدخلت دراسات التاريخ الطبيعي على نطاق واسع وإجراء المزيد من الدراساتفي 1970 و1980، مما أدى إلى زيادة انتشار استخدام العلاج الوقائي ضد الالتهابات الرئوية بين تدخلات أخرى. في عام 1972، الحائز على جائزة إيمي فيلم تلفزيوني بيل كوسبي، إلى كل أصدقائى على الشاطئ، يصور قصة والدي طفل يعاني من مرض hydroxycarbamide"هيدروكسي كاربامايد" الخلية المنجلية.[99] ورأى في 1990 تطوير ا ، وتقارير علاج من خلال يبدو زرع نخاع العظم في عام 2007.
بحث
- انظر أيضا: قائمة الباحثين مرض الخلية المنجلية
زرع دم الحبل السري
في ديسمبر 1998، أجرى باحثون من جامعة إيموري إجراء عملية زرع نخاع العظم التجريبية على مجموعة من 22 طفلا تقل أعمارهم عن 16 سنة.[100] واحد من هؤلاء المرضى، بن البالغ من العمر 12 عاما" كيوني بن"، كان على ما يبدو أول شخص يمكن علاجه من مرض الخلية المنجلية من خلال هذه الطريقة.[101] وكان مصدر الخلايا الجذعية من متبرع لا علاقة لها ب" بن". في 2007 مشروع قانون في مجلس الشيوخ جورجيا اقتراح جمع والتبرع بمواد الخلايا الجذعية، و"إنقاذ قانون شفاء"، والملقب ب "قانون في" تكريما له.
بحلول منتصف عام 2007 مجموعة مماثلة من التجارب السريرية في بالتيمور والشفاء أيضا العديد من البالغي.[102]
العلاج الجيني
في عام 2001 ذكر أن مرض فقر الدم المنجلي قد عولج بنجاح في الفئران باستخدام العلاج الجيني.[103][104] واستخدم الباحثون ناقلات فيروسية لجعل الفئران التي لديها أساسا نفس الخلل الذي يسبب إنتاج المنجل البشري يعبر عن إنتاج الهيموغلوبين الجيني HBF، الذي يتوقف-ينقطع عادة بشكل طبيعي في الافرد لإنتاج بعد فترة قصيرة من الولادة. في البشر، استخدام HBFهيدروكسي يوريا لتحفيز إنتاج كان من المعروف أن تقلل بشكل مؤقت من أعراض مرض الخلية المنجلية. أظهر الباحثون أن هذا الأسلوب العلاج الجيني هو وسيلة أكثر ديمومة لزيادة الإنتاج[105] وقد بدأت المرحلة 1 من التجارب السريرية العلاج الجيني لمرض فقر الدم المنجلي في البشر في عام 2014[106][107] اعتبارا من 2014 ولكن لم ترد تقارير عن تجارب عشوائية محكومة"مسيطر عليها" [108]
انظر أيضًا
المراجع
- "What Causes Sickle Cell Disease?"، National Heart, Lung, and Blood Institute.، 12 يونيو 2022، مؤرشف من الأصل في 27 فبراير 2021.
- "What Are the Signs and Symptoms of Sickle Cell Disease?"، National Heart, Lung, and Blood Institute.، 12 يونيو 2022، مؤرشف من الأصل في 9 يونيو 2022.
- "What Is Sickle Cell Disease?"، National Heart, Lung, and Blood Institute.، 12 يونيو 2022، مؤرشف من الأصل في 9 يونيو 2022.
- "How Is Sickle Cell Disease Diagnosed?"، National Heart, Lung, and Blood Institute.، 12 يونيو 2022، مؤرشف من الأصل في 27 فبراير 2021.
- "How Is Sickle Cell Disease Treated?"، National Heart, Lung, and Blood Institute.، 12 يونيو 2022، مؤرشف من الأصل في 9 مارس 2022.
- "Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015"، ncbi.nlm.nih.gov، 12 يونيو 2022، مؤرشف من الأصل في 23 مايو 2022.
- "Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015"، ncbi.nlm.nih.gov، 12 يونيو 2022، مؤرشف من الأصل في 23 مايو 2022.
- "About Sickle Cell Disease"، Genome.gov (باللغة الإنجليزية)، مؤرشف من الأصل في 2 أبريل 2022، اطلع عليه بتاريخ 12 يونيو 2022.
- "WHO | Sickle-cell disease and other haemoglobin disorders"، web.archive.org، 09 مارس 2016، اطلع عليه بتاريخ 12 يونيو 2022.
- "Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013"، ncbi.nlm.nih.gov، 12 يونيو 2022، مؤرشف من الأصل في 29 أبريل 2022.
- "Sickle cell disease"، thelancet، 12 يونيو 2022، مؤرشف من الأصل في 12 يونيو 2022.
- Oh؛ Stapleton؛ Whitley (29 أكتوبر 2011)، Textbook of Clinical Pediatrics (باللغة الإنجليزية)، Springer Science & Business Media، ISBN 978-3-642-02201-2، مؤرشف من الأصل في 14 يونيو 2022.
{{استشهاد بكتاب}}: الوسيط|مؤلف2=و|مؤلف-الأخير2=تكرر أكثر من مرة (مساعدة)، الوسيط|مؤلف=،|مؤلف1=، و|مؤلف-الأخير1=تكرر أكثر من مرة (مساعدة) - Savitt, T. L. (13 يناير 1989)، "Herrick's 1910 case report of sickle cell anemia. The rest of the story"، JAMA: The Journal of the American Medical Association، 261 (2): 266–271، doi:10.1001/jama.261.2.266، مؤرشف من الأصل في 25 يناير 2022.
- Serjeant, Graham R. (2010-12)، "One hundred years of sickle cell disease: Review"، British Journal of Haematology (باللغة الإنجليزية)، 151 (5): 425–429، doi:10.1111/j.1365-2141.2010.08419.x، مؤرشف من الأصل في 6 مايو 2022.
{{استشهاد بدورية محكمة}}: تحقق من التاريخ في:|تاريخ=(مساعدة) - Weatherall DJ, Clegg JB (2001)، "Inherited haemoglobin disorders: an increasing global health problem"، Bull. World Health Organ.، 79 (8): 704–12، PMC 2566499، PMID 11545326.
- Roberts I, de Montalembert M (يوليو 2007)، "Sickle cell disease as a paradigm of immigration hematology: new challenges for hematologists in Europe"، Haematologica، 92 (7): 865–71، doi:10.3324/haematol.11474، PMID 17606434، مؤرشف من الأصل في 2 أكتوبر 2011.
- GBD 2013 Mortality and Causes of Death (17 ديسمبر 2014)، "Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013."، Lancet، 385: 117–171، doi:10.1016/S0140-6736(14)61682-2، PMC 4340604، PMID 25530442.
- Wellems TE, Hayton K, Fairhurst RM (سبتمبر 2009)، "The impact of malaria parasitism: from corpuscles to communities"، J. Clin. Invest.، 119 (9): 2496–505، doi:10.1172/JCI38307، PMC 2735907، PMID 19729847، مؤرشف من الأصل في 14 يونيو 2018.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - WHO، "Sickle-cell anaemia - Report by the Secretariat" (PDF)، مؤرشف من الأصل (PDF) في 26 سبتمبر 2013، اطلع عليه بتاريخ 27 نوفمبر 2010.
- Aidoo M, Terlouw DJ, Kolczak MS, McElroy PD, ter Kuile FO, Kariuki S, Nahlen BL, Lal AA, Udhayakumar V (2002)، "Protective effects of the sickle cell gene against malaria morbidity and mortality"، Lancet، 359 (9314): 1311–2، doi:10.1016/S0140-6736(02)08273-9، PMID 11965279.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - National Heart, Lung and Blood Institute، "Sickle cell anemia, key points"، مؤرشف من الأصل في 6 أغسطس 2011، اطلع عليه بتاريخ 27 نوفمبر 2010.
- "September is Sickle Cell Awareness Month"، CDC، مؤرشف من الأصل في 30 يونيو 2017، اطلع عليه بتاريخ 06 فبراير 2011.
- "Sickle Cell Disease: Data & Statistics"، مراكز مكافحة الأمراض واتقائها، 16 سبتمبر 2011، مؤرشف من الأصل في 13 مايو 2019، اطلع عليه بتاريخ 08 نوفمبر 2011.
- American Academy of Pediatrics Section on Hematology/Oncology Committee on Genetics (2002)، "Health Supervision for Children with Sickle Cell Disease"، Pediatrics، 109 (3): 526–535، doi:10.1542/peds.109.3.526، PMID 11875155.
- Pass KA, Lane PA, Fernhoff PM, Hinton CF, Panny SR, Parks JS, Pelias MZ, Rhead WJ, Ross SI, Wethers DL, Elsas LJ (2000)، "US newborn screening system guidelines II: follow-up of children, diagnosis, management, and evaluation"، J Pediatr، 137 (37): S1–S46، doi:10.1067/mpd.2000.109437، PMID 11044838.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - "Sickle Cell Disease and Your Baby"، March of Dimes®، فبراير 2008، مؤرشف من الأصل في 28 يوليو 2018، اطلع عليه بتاريخ 11 نوفمبر 2014.
- Cinnchinsky EP, Mahoney DH, Landlaw SA (29 نوفمبر 2011)، "Uptodate: Sickle Cell Trait"، مؤرشف من الأصل في 20 يونيو 2017، اطلع عليه بتاريخ 08 نوفمبر 2011.
{{استشهاد ويب}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Bardakdjian J, Wajcman H (سبتمبر 2004)، "[Epidemiology of sickle cell anemia]"، Rev Prat (باللغة الفرنسية)، 54 (14): 1531–3، PMID 15558961.
- Bardakdjian-Michau J, Bahuau M, Hurtrel D, Godart C, Riou J, Mathis M, Goossens M, Badens C, Ducrocq R, Elion J, Perini JM (يناير 2009)، "Neonatal screening for sickle cell disease in France"، J. Clin. Pathol.، 62 (1): 31–3، doi:10.1136/jcp.2008.058867، PMID 19103855.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Le dépistage néonatal de la drépanocytose en France. Numéro thématique. La drépanocytose en France : des données épidémiologiques pour améliorer la prise en charge, Bardakdjian-Michau J, INVS, July 2012 نسخة محفوظة 31 يناير 2016 على موقع واي باك مشين.
- "Inheriting sickle cell anaemia - Live Well - NHS Choices"، www.nhs.uk، مؤرشف من الأصل في 26 يونيو 2016.
- "Who is offered screening and when?"، screening.nhs.uk، مؤرشف من الأصل في 31 ديسمبر 2014.
- "Give Blood - Resources - Sickle Cell and Blood Donation"، Give Blood، مؤرشف من الأصل في 23 أكتوبر 2015.
- "Why is Blood from Afro-Caribbean Donors Special?"، sicklecellsociety.org، مؤرشف من الأصل في 5 أبريل 2018.
- Jastaniah W (2011)، "Epidemiology of sickle cell disease in Saudi Arabia"، Annals of Saudi Medicine، 31 (3): 289–93، doi:10.4103/0256-4947.81540، PMC 3119971، PMID 21623060.
- Memish ZA, Saeedi MY (2011)، "Six-year outcome of the national premarital screening and genetic counseling program for sickle cell disease and β-thalassemia in Saudi Arabia"، Annals of Saudi Medicine، 31 (3): 229–35، doi:10.4103/0256-4947.81527، PMC 3119961، PMID 21623050.
- Awasthy N, Aggarwal KC, Goyal PC, Prasad MS, Saluja S, Sharma M (2008)، "Sickle cell disease: Experience of a tertiary care center in a nonendemic area"، Annals of Tropical Medicine and Public Health، 1 (1): 1–4، doi:10.4103/1755-6783.43069.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Life with sickle cell | Nation | Nepali Times نسخة محفوظة 22 أكتوبر 2017 على موقع واي باك مشين.
- Asnani MR, McCaw-Binns AM, Reid ME (2011)، "Excess Risk of Maternal Death from Sickle Cell Disease in Jamaica: 1998–2007"، PLoS ONE، 6 (10): e26281، doi:10.1371/journal.pone.0026281، PMC 3200316، PMID 22039456.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, Jordan L, Lanzkron SM, Lottenberg R, Savage WJ, Tanabe PJ, Ware RE, Murad MH, Goldsmith JC, Ortiz E, Fulwood R, Horton A, John-Sowah J (10 سبتمبر 2014)، "Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members."، JAMA، 312 (10): 1033–48، doi:10.1001/jama.2014.10517، PMID 25203083، مؤرشف من الأصل في 12 يونيو 2016.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - "BestBets: How long should an average sickle cell crisis last?"، مؤرشف من الأصل في 14 يونيو 2018، اطلع عليه بتاريخ 27 نوفمبر 2010.
- Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Aster, Jon (2009-05-28). Robbins and Cotran Pathologic Basis of Disease, Professional Edition: Expert Consult - Online (Robbins Pathology) (Kindle Locations 33498-33499). Elsevier Health. Kindle Edition.
- Olujohungbe A, Burnett AL (2013)، "How I manage priapism due to sickle cell disease"، British Journal of Haematology، 160 (6): 754–65، doi:10.1111/bjh.12199، PMID 23293942.
- Glassberg J (أغسطس 2011)، "Evidence-based management of sickle cell disease in the emergency department"، Emergency Medicine Practice، 13 (8): 1–20, quiz 20، PMID 22164362.
- Anie KA, Green J (2012)، Anie, Kofi A (المحرر)، "Psychological therapies for sickle cell disease and pain"، Cochrane Database of Systematic Reviews (Online)، 2: CD001916، doi:10.1002/14651858.CD001916.pub2، PMID 22336781.
- Pearson HA (أغسطس 1977)، "Sickle cell anemia and severe infections due to encapsulated bacteria"، J Infect Dis، 136 Suppl: S25–30، doi:10.1093/infdis/136.Supplement.S25، ISSN 0022-1899، PMID 330779، مؤرشف من الأصل (Free full text) في 5 يوليو 2016.
- Wong WY, Powars DR, Chan L, Hiti A, Johnson C, Overturf G (مارس 1992)، "Polysaccharide encapsulated bacterial infection in sickle cell anaemia: a thirty year epidemiologic experience"، Am J Hematol، 39 (3): 176–82، doi:10.1002/ajh.2830390305، PMID 1546714.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Khatib R, Rabah R, Sarnaik SA (يناير 2009)، "The spleen in the sickling disorders: an update"، Pediatric Radiology، 39 (1): 17–22، doi:10.1007/s00247-008-1049-9، PMID 19002450.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Glassberg (أغسطس 2011)، "Evidence-based management of sickle cell disease in the emergency department."، Emergency medicine practice، 13 (8): 1–20, quiz 20، PMID 22164362.
- Mekontso Dessap A, Leon R, Habibi A, Nzouakou R, Roudot-Thoraval F, Adnot S, Godeau B, Galacteros F, Brun-Buisson C, Brochard L, Maitre B (2008)، "Pulmonary hypertension and cor pulmonale during severe acute chest syndrome in sickle cell disease"، Am. J. Respir. Crit. Care Med.، 177 (6): 646–53، doi:10.1164/rccm.200710-1606OC، PMID 18174543.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Paul RN, Castro OL, Aggarwal A, Oneal PA (2011)، "Acute chest syndrome: sickle cell disease"، Eur. J. Haematol.، 87 (3): 191–207، doi:10.1111/j.1600-0609.2011.01647.x، PMID 21615795.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Aster, Jon (2009-05-28). Robbins and Cotran Pathologic Basis of Disease, Professional Edition: Expert Consult - Online (Robbins Pathology) (Kindle Location 33329). Elsevier Health. Kindle Edition.
- Slavov SN, Kashima S, Pinto AC, Covas DT (أغسطس 2011)، "Human parvovirus B19: general considerations and impact on patients with sickle-cell disease and thalassemia and on blood transfusions"، FEMS Immunology and Medical Microbiology، 62 (3): 247–62، doi:10.1111/j.1574-695X.2011.00819.x، PMID 21585562.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Balgir RS (مارس 2012)، "Community expansion and gene geography of sickle cell trait and G6PD deficiency, and natural selection against malaria: experience from tribal land of India"، Cardiovascular & Hematological Agents in Medicinal Chemistry، 10 (1): 3–13، doi:10.2174/187152512799201190، PMID 22264009.
- Jadavji T, Prober CG (أبريل 1985)، "Dactylitis in a child with sickle cell trait"، Can Med Assoc J، 132 (7): 814–5، ISSN 0008-4409، PMC 1345873، PMID 3978504.
- Worrall VT, Butera V (1976)، "Sickle-cell dactylitis"، J Bone Joint Surg Am، 58 (8): 1161–3، PMID 1002763، مؤرشف من الأصل في 23 سبتمبر 2016.
- Miller ST (مايو 2011)، "How I treat acute chest syndrome in children with sickle cell disease"، Blood، 117 (20): 5297–305، doi:10.1182/blood-2010-11-261834، PMID 21406723.
- Green NS, Fabry ME, Kaptue-Noche L, Nagel RL (أكتوبر 1993)، "Senegal haplotype is associated with higher HbF than Benin and Cameroon haplotypes in African children with sickle cell anemia"، Am. J. Hematol.، 44 (2): 145–6، doi:10.1002/ajh.2830440214، ISSN 0361-8609، PMID 7505527.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Allison AC (أكتوبر 2009)، "Genetic control of resistance to human malaria"، Current Opinion in Immunology، 21 (5): 499–505، doi:10.1016/j.coi.2009.04.001، PMID 19442502.
- Kwiatkowski DP (أغسطس 2005)، "How Malaria Has Affected the Human Genome and What Human Genetics Can Teach Us about Malaria"، Am. J. Hum. Genet.، 77 (2): 171–92، doi:10.1086/432519، ISSN 0002-9297، PMC 1224522، PMID 16001361.
- Ponçon N, Toty C, L'Ambert G, Le Goff G, Brengues C, Schaffner F, Fontenille D (2007)، "Biology and dynamics of potential malaria vectors in Southern France"، Malar. J.، 6 (1): 18، doi:10.1186/1475-2875-6-18، PMC 1808464، PMID 17313664.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Lesi FE, Bassey EE (يوليو 1972)، "Family study in sickle cell disease in Nigeria"، J Biosoc Sci، 4 (3): 307–13، doi:10.1017/S0021932000008622، PMID 5041262.
- "How Does Sickle Cell Cause Disease?"، مؤرشف من الأصل في 16 أكتوبر 2018، اطلع عليه بتاريخ 27 نوفمبر 2010.
- "Sickle Cell Anemia: eMedicine Emergency Medicine"، مؤرشف من الأصل في 4 ديسمبر 2010، اطلع عليه بتاريخ 27 نوفمبر 2010.
- Clarke GM, Higgins TN (أغسطس 2000)، "Laboratory investigation of hemoglobinopathies and thalassemias: review and update"، Clin. Chem.، 46 (8 Pt 2): 1284–90، PMID 10926923، مؤرشف من الأصل في 28 نوفمبر 2011.
- "BestBets: Does routine urinalysis and chest radiography detect occult bacterial infection in sickle cell patients presenting to the accident and emergency department with painful crisis?"، مؤرشف من الأصل في 14 يونيو 2018، اطلع عليه بتاريخ 27 نوفمبر 2010.
- Lee, C., Davies, S.,& Dezatoux, C. (2000). Neonatal Screening for sickle cell disease. The Cochrane Collaboration. John Wiley & Sons, Ltd.
- Oniyangi O, Omari AA (2006)، Oniyangi, Oluseyi (المحرر)، "Malaria chemoprophylaxis in sickle cell disease"، Cochrane Database of Systematic Reviews، 13 (4): CD003489، doi:10.1002/14651858.CD003489.pub2، PMID 17054173.
- Aldrich TK, Nagel RL. (1998)، "Pulmonary Complications of Sickle Cell Disease."، في Reynolds HY, Bone RC, Dantzker DR, George RB, Matthay RA (المحرر)، Pulmonary and Critical Care Medicine (ط. 6th)، St. Louis: Mosby، ص. 1–10، ISBN 0-8151-1371-4.
{{استشهاد بكتاب}}: صيانة CS1: أسماء متعددة: قائمة المحررون (link) - Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR (مايو 1995)، "Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia"، N. Engl. J. Med.، 332 (20): 1317–22، doi:10.1056/NEJM199505183322001، ISSN 0028-4793، PMID 7715639.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, Orringer E, Bellevue R, Olivieri N, Eckman J, Varma M, Ramirez G, Adler B, Smith W, Carlos T, Ataga K, DeCastro L, Bigelow C, Saunthararajah Y, Telfer M, Vichinsky E, Claster S, Shurin S, Bridges K, Waclawiw M, Bonds D, Terrin M (أبريل 2003)، "Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment"، JAMA، 289 (13): 1645–51، doi:10.1001/jama.289.13.1645، PMID 12672732، مؤرشف من الأصل في 2 نوفمبر 2010.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Platt OS (مارس 2008)، "Hydroxyurea for the treatment of sickle cell anemia"، N. Engl. J. Med.، 358 (13): 1362–9، doi:10.1056/NEJMct0708272، PMID 18367739.
- Drasar E, Igbineweka N, Vasavda N, Free M, Awogbade M, Allman M, Mijovic A, Thein SL (مارس 2011)، "Blood transfusion usage among adults with sickle cell disease - a single institution experience over ten years"، Br. J. Haematol.، 152 (6): 766–70، doi:10.1111/j.1365-2141.2010.08451.x، PMID 21275951.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Gyang E, Yeom K, Hoppe C, Partap S, Jeng M (يناير 2011)، "Effect of chronic red cell transfusion therapy on vasculopathies and silent infarcts in patients with sickle cell disease"، Am. J. Hematol.، 86 (1): 104–6، doi:10.1002/ajh.21901، PMID 21117059.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Mirre E, Brousse V, Berteloot L, Lambot-Juhan K, Verlhac S, Boulat C, Dumont MD, Lenoir G, de Montalembert M (مارس 2010)، "Feasibility and efficacy of chronic transfusion for stroke prevention in children with sickle cell disease"، Eur. J. Haematol.، 84 (3): 259–65، doi:10.1111/j.1600-0609.2009.01379.x، PMID 19912310.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Walters MC, Patience M, Leisenring W, Eckman JR, Scott JP, Mentzer WC, Davies SC, Ohene-Frempong K, Bernaudin F, Matthews DC, Storb R, Sullivan KM (أغسطس 1996)، "Bone marrow transplantation for sickle cell disease"، N. Engl. J. Med.، 335 (6): 369–76، doi:10.1056/NEJM199608083350601، PMID 8663884.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Wierenga KJ, Hambleton IR, Lewis NA (2001)، "Survival estimates for patients with homozygous sickle-cell disease in Jamaica: A clinic-based population study"، Lancet، 357 (9257): 680–683، doi:10.1016/s0140-6736(00)04132-5، PMID 11247552.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Aster, Jon (2009-05-28). Robbins and Cotran Pathologic Basis of Disease, Professional Edition: Expert Consult - Online (Robbins Pathology) (Kindle Locations 33530-33531). Elsevier Health. Kindle Edition.
- Kavanagh PL, Sprinz PG, Vinci SR, Bauchner H, Wang CJ (2011)، "Management of children with sickle cell disease: a comprehensive review of the literature"، Pediatrics، 128 (6): e1552–74، doi:10.1542/peds.2010-3686، PMID 22123880، مؤرشف من الأصل في 18 أكتوبر 2018.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Adams RJ, Ohene-Frempong K, Wang W (2001)، "Sickle cell and the brain"، Hematology Am Soc Hematol Educ Program، 2001 (1): 31–46، doi:10.1182/asheducation-2001.1.31، PMID 11722977، مؤرشف من الأصل في 3 أكتوبر 2011.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Adams RJ (نوفمبر 2007)، "Big strokes in small persons"، Arch. Neurol.، 64 (11): 1567–74، doi:10.1001/archneur.64.11.1567، PMID 17998439، مؤرشف من الأصل في 2 أبريل 2012.
- Martí-Carvajal؛ Dunlop؛ Agreda-Perez (18 أكتوبر 2004)، "Treatment for avascular necrosis of bone in people with sickle cell disease."، The Cochrane database of systematic reviews (4): CD004344، doi:10.1002/14651858.CD004344.pub2، PMID 15495103.
- Kenny MW, George AJ, Stuart J (يوليو 1980)، "Platelet hyperactivity in sickle-cell disease: a consequence of hyposplenism"، Journal of Clinical Pathology، 33 (7): 622–5، doi:10.1136/jcp.33.7.622، PMC 1146172، PMID 7430367، مؤرشف من الأصل في 01 أبريل 2020، اطلع عليه بتاريخ 23 مارس 2011.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Chrouser KL, Ajiboye OB, Oyetunji TA, Chang DC (أبريل 2011)، "Priapism in the United States: the changing role of sickle cell disease"، American Journal of Surgery، 201 (4): 468–74، doi:10.1016/j.amjsurg.2010.03.017، PMID 21421100.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Almeida A, Roberts I (مايو 2005)، "Bone involvement in sickle cell disease"، Br. J. Haematol.، 129 (4): 482–90، doi:10.1111/j.1365-2141.2005.05476.x، PMID 15877730، مؤرشف من الأصل في 01 أبريل 2020.
- Rudge FW (1991)، "Hyperbaric oxygen therapy in the treatment of sickle cell leg ulcers"، J. Hyperbaric Med، 6 (1): 1–4، مؤرشف من الأصل في 01 أبريل 2020، اطلع عليه بتاريخ 23 مارس 2011.
- Elagouz M, Jyothi S, Gupta B, Sivaprasad S (يوليو 2010)، "Sickle cell disease and the eye: old and new concepts"، Survey of Ophthalmology، 55 (4): 359–77، doi:10.1016/j.survophthal.2009.11.004، PMID 20452638، مؤرشف من الأصل في 8 سبتمبر 2018، اطلع عليه بتاريخ 23 مارس 2011.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Caughey MC, Poole C, Ataga KI, Hinderliter AL (09 أبريل 2015)، "Estimated pulmonary artery systolic pressure and sickle cell disease: a meta-analysis and systematic review"، British Journal of Haematology، 170: 416–424، doi:10.1111/bjh.13447.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Powars DR, Elliott-Mills DD, Chan L, Niland J, Hiti AL, Opas LM, Johnson C (أكتوبر 1991)، "Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality"، Annals of Internal Medicine، 115 (8): 614–20، doi:10.7326/0003-4819-115-8-614، ISSN 0003-4819، PMID 1892333.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Lebby R (1846)، "Case of absence of the spleen"، Southern J of Med Pharmacol، 1: 481–3.
- Ballas SK, Gupta K, Adams-Graves P (1 نوفمبر 2012)، "Sickle cell pain: a critical reappraisal."، Blood، 120 (18): 3647–56، doi:10.1182/blood-2012-04-383430، PMID 22923496، مؤرشف من الأصل في 12 أكتوبر 2018.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Herrick JB (1910)، "Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia"، Arch. Intern. Med.، 6 (5): 517–521، doi:10.1001/archinte.1910.00050330050003، مؤرشف من الأصل في 24 فبراير 2012.; reprinted as Herrick JB (2001)، "Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. 1910"، The Yale journal of biology and medicine، 74 (3): 179–84، PMC 2588723، PMID 11501714، مؤرشف من الأصل في 24 فبراير 2012.
- Savitt TL, Goldberg MF (يناير 1989)، "Herrick's 1910 case report of sickle cell anemia. The rest of the story"، JAMA، 261 (2): 266–71، doi:10.1001/jama.261.2.266، PMID 2642320.
- Serjeant GR (ديسمبر 2010)، "One hundred years of sickle cell disease."، British journal of haematology، 151 (5): 425–9، doi:10.1111/j.1365-2141.2010.08419.x، PMID 20955412، مؤرشف من الأصل في 5 أغسطس 2017.
- Washburn, R.E. (1911)، "Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia"، The Virginia Medical Semi-Monthly، 15 (21): 490–493.
- "UVa Hospital Celebrating 100 Years"، University of Virginia، مؤرشف من الأصل في 10 أكتوبر 2018، اطلع عليه بتاريخ 28 يناير 2015.
- Mason VR (1922)، "Sickle cell anemia"، JAMA، 79 (14): 1318–1320، doi:10.1001/jama.254.14.1955، PMID 3900438.
- Pauling L, Itano HA (1949)، "Sickle cell anemia, a molecular disease"، Science، 110 (2865): 543–548، doi:10.1126/science.110.2865.543، PMID 15395398.
- "sickle+cell+anemia"&SearchStyle=&dTitle=Foster,+Gloria&TabRecordType=Biography&BioCountPass=11&SubCountPass=3&DocCountPass=0&ImgCountPass=1&MapCountPass=0&FedCountPass=&MedCountPass=1&NewsCountPass=0&RecPosition=10&AmericanData=&WomenData=&AFHCData=Set&IndianData=&WorldData=&AncientData=&GovernmentData= "Foster, Gloria"، Facts On File History Database، مؤرشف من "sickle+cell+anemia"&SearchStyle=&dTitle=Foster,+Gloria&TabRecordType=Biography&BioCountPass=11&SubCountPass=3&DocCountPass=0&ImgCountPass=1&MapCountPass=0&FedCountPass=&MedCountPass=1&NewsCountPass=0&RecPosition=10&AmericanData=&WomenData=&AFHCData=Set&IndianData=&WorldData=&AncientData=&GovernmentData= الأصل في 5 مارس 2016، اطلع عليه بتاريخ 25 فبراير 2015.
- MMS: Error نسخة محفوظة 19 أكتوبر 2017 على موقع واي باك مشين.
- Keone Penn, 27: Medical trailblazer wanted to be a chef | www.ajc.com نسخة محفوظة 04 مارس 2016 على موقع واي باك مشين.
- Breakthrough: Baltimore woman becomes one of the first adults to be cured of sickle-cell disease - Winston-Salem Journal: Archives نسخة محفوظة 02 يوليو 2017 على موقع واي باك مشين.
- Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, Bouhassira EE, Acharya SA, Ellis J, London IM, Eaves CJ, Humphries RK, Beuzard Y, Nagel RL, Leboulch P (2001)، "Correction of Sickle Cell Disease in Transgenic Mouse Models by Gene Therapy"، Science، 294 (5550): 2368–71، doi:10.1126/science.1065806، PMID 11743206.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Wilson, Jennifer Fisher (18 مارس 2002)، "Murine Gene Therapy Corrects Symptoms of Sickle Cell Disease"، The Scientist – Magazine of the Life Sciences، مؤرشف من الأصل في 01 أبريل 2020، اطلع عليه بتاريخ 17 ديسمبر 2014.
- St. Jude Children's Research Hospital (04 ديسمبر 2008)، "Gene Therapy Corrects Sickle Cell Disease In Laboratory Study"، ScienceDaily، مؤرشف من الأصل في 13 يونيو 2018، اطلع عليه بتاريخ 17 ديسمبر 2014.
- (15 December 2014) Stem Cell Gene Therapy for Sickle Cell Disease, ClinicalTrials.gov Identifier: NCT02247843 ClinicalTrials.gov, U.S. National Institutes of Health, Retrieved 17 December 2014 نسخة محفوظة 24 أبريل 2017 على موقع واي باك مشين.
- (15 December 2014) "gene+therapy"+OR+"gene+transfer"+OR+"virus+delivery"&recr=Open&lup_s=11/17/2014&lup_d=30 Collection and Storage of Umbilical Cord Stem Cells for Treatment of Sickle Cell Disease; ClinicalTrials.gov Identifier: NCT00012545 ClinicalTrials.gov, U.S. National Institutes of Health, Retrieved 17 December 2014 نسخة محفوظة 17 ديسمبر 2019 على موقع واي باك مشين.
- Olowoyeye؛ Okwundu (10 أكتوبر 2014)، "Gene therapy for sickle cell disease."، The Cochrane database of systematic reviews، 10: CD007652، doi:10.1002/14651858.CD007652.pub4، PMID 25300171.
 بوابة طب
بوابة طب