ورم حميد
الوَرَم الحَميد (بالإنجليزية: Benign Tumor) هو مرض خلوي ينتج عن تجمع خلايا تتكاثر في مواضع معينة في الجسم، وتتميز بمحدودية تطورها ومشابهتها لخلايا عضو المنشأ، بالإضافة لبطئ نموها وعدم انتشار الورم إلى الأعضاء الأخرى عكس الورم الخبيث.[1][2][3] هناك بعض أنواع الأورام الحميدة قد تتحول إلى أورام خبيثة مثل الورم الغدي النبيبي (أحد أنواع سرطان القولون) من خلال عملية تعرف باسم تطور الورم.[4] معظم الأورام الحميدة لا تحتاج للعلاج وإن كانت له حاجة، فسيقتصر على الجراحة. عادة ما تكون محاطة بسطح خارجي (غمد ليفي من النسيج الضام) أو تبقى محتواة داخل النسيج الطلائي.[5] تشمل الأمثلة الشائعة للأورام الحميدة الشامات والأورام الليفية الرحمية.
| ورم حميد | |
|---|---|
| معلومات عامة | |
| الاختصاص | علم الأمراض |
| التاريخ | |
| وصفها المصدر | الموسوعة السوفيتية الكبرى |

سرعه نموها عادة بطيئة جدا وطريقة تحركها بالتوسع في نفس العضو وغالبا ما تكون محلية أي أنها لا تنتقل من عضو لأخر ولكن هذا لا يمنع تأثيرها على الأعضاء المجاورة، وإذا تم استئصالها تمامًا فإنها عادة لا ترجع مرة أخرى بعكس الأورام الخبيثة.
يمكن أن تؤثر على المريض في الحالات التالية:
- إذا نمت في مكان تضغط فيه على عضو مهم وحساس مثل المخ أو النخاع الشوكي.
- إذا نمت في مكان تضغط فيه على عضو رخو مثل الكبد أو الأمعاء فانها يمكن ان تتسبب في إيقاف عمل هذا العضو أو انسداد الامعاء.
- إذا كانت موجودة بجانب إحدى الغدد الصماء في الجسم فيمكن ان تضغط على الغدة وإما ان تزيد افرازها أو تقلله حسب الحالات.
- بالإضافة إلى إمكانية تحولها إلى ورم خبيث.
العلامات والأعراض
الأورام الحميدة متنوعة للغاية وقد تكون غير مصحوبة بأعراض أو قد تسبب أعراضاً معينة حسب موقعها التشريحي ونوع النسيج. تنمو الأورام الحميدة نحو الخارج وتنتج كتلاً كبيرة مستديرة والتي يمكن أن تسبب ما يعرف باسم «تأثير الكتلة (mass effect)». هذا النمو يمكن أن يشكل ضغطاً على الأنسجة أو الأعضاء المحلية والذي من الممكن أن يسبب العديد من التأثيرات كانسداد القنوات، إنخفاض تدفق الدم، موت الأنسجة والألم العصبي أو تلف الأعصاب.[6] بعض الأورام تنتج أيضا هرمونات يمكن أن تؤدي إلى حالات تهدد الحياة. الأورام الجزيرية يمكنها إنتاج كميات كبيرة من الأنسولين مما يؤدي إلى نقص السكر في الدم.[7][8] أورام الغدة النخامية يمكن أن تسبب ارتفاعا في مستويات العديد من الهرمونات مثل:[9] هرمون النمو وعامل النمو شبيه الانسولين-1 والذي يؤدي إلى ضخامة الأطراف، الهرمون الموجه لقشر الكظر وكورتيزول يؤدي إلى مرض كوشينغ، هرمون منبه الدرقية والذي يتسبب في فرط الدرقية والهرمون المنشط للحوصلة والهرمون المنشط للجسم الأصفر وبرولاكتين. انغلاف الأمعاء يمكن أن يحدث مع العديد من الأورام القولونية الحميدة.[10] يمكن أن تحدث آثار تجميلية بسبب الأورام وخاصةً تلك الموجودة في الجلد، مما قد يسبب آثارًا نفسية على الشخص المصاب بالورم.[11] يمكن للأورام الوعائية أن تنزف وفي بعض الحالات يكون النزيف حادا مما يؤدي إلى فقر الدم.[12]
الأسباب
متلازمة الورم الدموي
تتكون متلازمة الورم الدموي PTEN من أربعة اضطرابات عابية مميزة تتميز بطفرات جينية في جين PTEN ؛ متلازمة كاودن، متلازمة بانيان- رايلي- روفالكابا، متلازمة بروتيوس ومتلازمة بروتيوس الشبيهة. على الرغم من أن لديهم جميعًا سمات سريرية مميزة، إلا أن تكوين الأورام الوعائية يحدث في جميع المتلازمات الأربعة. PTEN هو جين مثبط للورم يشارك في الإشارات الخلوية. يسمح بروتين PTEN الغائب أو المختل وظيفيًا للخلايا بالتكاثر المفرط، مما يتسبب في أورام عقيمة.[13]
متلازمات أخرى
متلازمة كاودن هي جسمية مهيمنة اضطراب وراثي يتميز حميدة متعددة هامارتوما (داء المشعرات ومخاطي جلدي الورم الحليمي حطاطات) فضلا عن الاستعداد لسرطانات أجهزة متعددة بما في ذلك الثدي والغدة الدرقية.[14][15] متلازمة بانيان- رايلي- روفالكابا هي اضطراب خلقي ومتلازمة فرط نمو نادرة ومرض مسبب للأورام العيبية. تتضمن المتلازمة تشكل أورام شحمية متعددة تحت الجلد وضخامة رأس وأورام وعائية دموية. كبر الرأس، وبقع في حشفة القضيب.[13][16] متلازمة بروتيوس يتميز الشامات، فرط غير المتماثلة من مختلف أجزاء الجسم، والدهنية التقلبات الأنسجة، ورم غدي كيسي، أورام، تشوه الأوعية الدموية.[17][18]
داء السلائل الورمي الغدي العائلي

داء السلائل الورمي الغدي العائلي (FAP) هو متلازمة سرطانية عائلية ناتجة عن طفرات في جين APC. في هذا الاضطراب، توجد الأورام الحميدة الغدية في القولون والتي ستتطور إلى سرطان القولون ما لم تتم إزالتها.[19] الجين APC هو مثبط للورم. يشارك منتجها البروتيني في العديد من العمليات الخلوية. يؤدي تعطيل جين APC إلى تراكم بروتين يسمى-بيتا-كاتينين، والذي ينشط عاملين للنسخ: عامل الخلايا التائية (TCF) وعامل المُحسِّن اللمفاوي (LEF). هذه تسبب انتفاخ العديد من الجينات المشاركة في تكاثر الخلايا والتمايز والهجرة والاستماتة (موت الخلايا المبرمج)، مما يتسبب في نمو الأورام الحميدة [20]
التصلب الحدبي
تصلب حدبي (TSC) هو وراثي اضطراب وراثي المهيمنة التي تسببها طفرات في الجينات TSC1 وTSC2، والتي تنتج البروتينات TSC1 وتوبرين، على التوالي. يتظاهر هذا الاضطراب مع العديد من الأورام الوعائية الحميدة بما في ذلك الأورام الليفية الوعائية، والأورام الوعائية الوعائية الكلوية، والورم العضلي الوعائي اللمفاوي الرئوي. يعمل كل من توبرين وTSC1 على تثبيط بروتين mTOR في فسيولوجيا الخلية الطبيعية، كما يؤدي تعطيل مثبطات الورم TSC إلى زيادة نشاط mTOR. وهذا يؤدي إلى تنشيط الجينات وإنتاج البروتينات التي تزيد من نمو الخلايا.[21][22][23]
داء فون هيبل - لينداو
مرض فون هيبل لينداو هو متلازمة سرطانية وراثية بشكل كبير تزيد بشكل كبير من خطر الإصابة بأورام مختلفة بما في ذلك الأورام الأرومية ورم وعائي الحميدة وورم القواتم الخبيث وسرطان الخلايا الكلوية وأورام الغدد الصماء البنكرياسية وأورام الكيس اللمفي. وهو ناتج عن طفرات جينية في جين مثبط ورم فون هيبل - لينداو. يشارك بروتين VHL (pVHL) في الإشارات الخلوية في الخلايا المتعطشة للأكسجين (نقص أكسجة الورم). يتمثل أحد أدوار pVHL في التسبب في التدهور الخلوي لبروتين آخر، HIF1A [الإنجليزية]. يؤدي pVHL المختل وظيفيًا إلى تراكم HIF1α، والذي ينشط العديد من الجينات المسؤولة عن إنتاج المواد المشاركة في نمو الخلايا وإنتاج الأوعية الدموية: VEGF وPDGFβ وTGFα وإريثروبويتين.[24]
متلازمة ستيرج - ويبر
هو متلازمة نادرة تصيب الدماغ وجلد الرأس والعين. حيث تكون على شكل تشوه في الشعيرات الدموية في الوجه (وحمة نبيذية خمرية) في المنطقة الجلدية التي يغذيها العصب العيني عادة (V1) من العصب الثلاثي التوائم، ولكن ليست جميع الحالات يصاحبها الوحمة الخمرية. قد تكون الوحمة في أماكن أخرى من جلد الوجه كالمنطقة التي يغذيها العصب الفكي العلوي أو السفلي.
العديد من المرضى الذين يعانون من الوحمة الخمرية يصاحبها زيادة في ضغط العين التي في نفس منطقة الوحمة أو الجلوكوما، كما أنهم يعانون من تشوه وريدي دماغي (ورم وعائي سحائي)، حيث أن ذلك يؤدي إلى تجمعات كلسية في قشرة الدماغ مما يؤدي إلى فقدان أو ضمور في الأنسجة العصبية. التلف في الأنسجة العصبية يظهر على المريض كصداع، أو تشنجات عصبية، أو نوبات تشبه السكتة الدماغية، أو شلل نصفي، أو خلل في المجال البصري، أو ضعف ادراكي، أو صعوبات في التعلم، وقد يعاني المريض من أكثر من عرض بنفس الوقت.

يعتمد التشخيص سريرياً على وجود عرضين من الثلاثة السابق ذكرهم وهم: الوحمة الخمرية، والجلوكوما، والورم الوعائي السحائي.
تم الإبلاغ مؤخراً من قبل عدد من الأطروحات العلمية عن وجود علاقة بين الطفرة الجينية الجسدية GNAQ على الكروموسوم 9q21 كسبب للمتلازمة.
عند تشخيص المريض سريرياً فانه ينبغي إجراء صورة رنين مغناطيسي للدماغ، واجراء تخطيط لكهربية الدماغ لمعرفة ان كان هنالك نشاط كهربي غير طبيعي للتعامل معه، واجراء فحص للعين وخاصة في الجهة المصابة، واجراء تقييم نفسي سلوكي للمريض.
يحتاج المريض لعلاج دوائي أو جراحي اعتماداً على الأعراض التي يشتكي منها حيث أنه في بعض الأحيان قد يحتاج لتداخل جراحي لازالة المنطقة من الدماغ التي تعاني من شحنات غير طبيعية، وقد يحتاج المريض لازالة جهة كاملة من الدماغ إذا كانت الأعراض شديدة جداً لا تستجيب للدواء. [25]
ورم عصبي ليفي (النوع الأول)
(بالإنجليزية: Neurofibromatosis type I)، هو مرض وراثي جسمي سائد نتيجة لطفرة في الجين NF1 المتواجد في كروموسوم 17 (17q11.2) حيث أن هذا الجين يعد المنشأ للبروتين الليفي العصبي (بالإنجليزية: neurofibromin)، وفي حال وجود خلل في هذا البروتين يؤدي ذلك إلى الورم العصبي الليفي الأول حيث تظهر الأعراض حسب مقدار الطفرة المصاحبة للجين. ان نوع الطفرة ومقدارها لا يلزم أن يكون متشابه بين المورث والوارث، وهذا يفسر اختلاف الأعراض المرضية وحدتها بين المورث (كالأب مثلاً)، وبين الوارث (كالابن).
ان هذا المرض يظهر في عدد من الأجهزة الجسمانية، ويصيب تقريباً شخص من كل 3500 شخص، حيث أن ظهور الأعراض للشخص الحامل للطفرة في العادة يكون في بداية العمر، وتزداد احتمالية ظهورها مع العمر لتصل إلى 100% على عمر 20 سنة.
يتم تشخيص المرض سريرياً بظهور عرضين على الأقل من الأعراض السبعة التالية:
- ظهور 6 بقع أو أكثر على الجلد من النمش المسمى مجازاً (بقعة القهوة مع الحليب) (بالفرنسية: Café au lait)، على أن يزيد قطرها عن 5 ملم في المرضى قبل سن البلوغ، وعن 15 ملم في المرضى بعد سن البلوغ.
- اثنان أو أكثر من الورم العصبي الليفي من أي نوع أو واحد من الورم الليفي العصبي الضفيري.


- نمش في منطقة الإبط (علامة كرو) أو المناطق الأربية.
- الورم الدبقي البصري.
- عقيدات ليش (اثنتان أو أكثر)، وهي ورم قزحي مصطبغ.
- خلل التنسج الليفي في العظم الوتدي، أو ترقق القشرة العظمية في العظام الطويلة.
- قريب من الدرجة الأولى يعاني من ورم العصب الليفي الأول.
يتم إجراء الفحص الجيني للأشخاص الذين يتم الشك في حملهم للمرض لكن لا تظهر عليهم الأعراض بالصورة النمطية السابقة، أو يظهر عليهم عرض واحد فقط من الأعراض السابقة، أو للجنين في حال رغبة الوالدين بمعرفة إذا كان طفلهم يحمل المرض، أو للأشخاص الذين تظهر عليهم أعراض المرض لكن لا يوجد لديهم أقرباء من الدرجة الأولى يعانون منه، أو للتفريق بين المرضى الذين يعانون من أمراض شبيهة بالورم العصبي الليفي.
ان الأعراض المصاحبة للورم العصبي الليفي النوع الأول لا تقتصر على ما سبق، بل قد تشمل أيضاً العديد من الأورام الحميدة في الجهاز الهضمي، أو خلل عظمي نسيجي، أو خلل قلبي وعائي.[26]
ورم عصبي ليفي (النوع الثاني)
(بالإنجليزية: Neurofibromatosis type II)، هو مرض وراثي جسمي سائد نتيجة لطفرة في الجين المثبط للورم NF2 المتواجد في كروموسوم 22 (22q12)، يصيب هذا المرض تقريباً شخص من كل 25000 شخص، ونسبة ظهور أعراضه للشخص حامل الجين المصاب تزداد مع تقدم العمر لتصل إلى 100% على عمر 60 سنة. نصف المرضى الذين يتم تشخيصهم بالمرض يظهر لديهم بدون أن يكون هنالك علاقة وراثية مباشرة مع شخص مصاب، على أن الشخص المصاب يورث المرض لأبنائه كصفة سائدة.
يتم تشخيص المرض سريرياً عند وجود أي نقطة مما يلي:
- ورم شفاني دهليزي على الجهتين أو ورم شفاني دهليزي على جهة واحدة مصاحب لفرد في العائلة مصاب بالمرض أو الإصابة باثنان من: ورم سحائي، ورم دبقي، ورم ليفي عصبي، ورم شفاني، عتامة الجزء الخلفي تحت كابسولة عدسة العين.
- ورم شفاني دهليزي على جهة واحدة بالإضافة للإصابة باثنان من: ورم سحائي، ورم دبقي، ورم ليفي عصبي، ورم شفاني، عتامة الجزء الخلفي تحت كابسولة عدسة العين.
- الإصابة بورمين سحائيين بمكانين مختلفين بالإضافة إلى: الإصابة بورم شفاني دهليزي أو الإصابة باثنين من: ورم دبقي، ورم شفاني، ومرض الساد.
ان الورم العصبي الليفي الثاني يعد سبباً للإصابة بالعديد من الأورام في الجهاز العصبي المركزي السابق ذكرها ويتم العلاج جراحياً أو اشعاعياً الورم الذي يؤدي إلى حدوث أعراض مؤثرة على النشاط الطبيعي اليومي للمريض.[27]
الآلية

حميد ضد خبيث
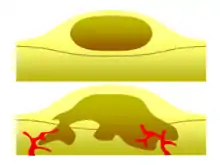
أحد أهم العوامل في تصنيف الورم على أنه ورم حميد أو خبيث هو قدرته على التوغل. إذا كان الورم يفتقر إلى القدرة على غزو الأنسجة المجاورة أو الانتشار إلى مواقع بعيدة عن طريق الانتقال فإنه يكون حميدًا، في حين أن الأورام الغازية أو النقيلية تكون خبيثة.[28] لهذا السبب، لا يتم تصنيف الأورام الحميدة على أنها سرطانية.[29] تنمو الأورام الحميدة في منطقة محصورة عادة ما تكون مغلفة في كبسولة نسيج ضام ليفي. تختلف معدلات نمو الأورام الحميدة والخبيثة أيضًا؛ تنمو الأورام الحميدة بشكل أبطأ من الأورام الخبيثة. على الرغم من أن الأورام الحميدة تشكل مخاطر صحية أقل من الأورام الخبيثة، إلا أن كلاهما يمكن أن يهدد الحياة في مواقف معينة. هناك العديد من الخصائص العامة التي تنطبق على الأورام الحميدة أو الخبيثة، ولكن في بعض الأحيان قد يظهر نوع واحد خصائص الآخر. على سبيل المثال، غالبًا ما تكون الأورام الحميدة متمايزة جيدًا وغالبًا ما تكون الأورام الخبيثة غير متمايزة. ومع ذلك، يمكن أن تحدث أورام حميدة غير متمايزة وأورام خبيثة متمايزة.[30][31] على الرغم من أن الأورام الحميدة تنمو ببطء بشكل عام، فقد تم أيضًا توثيق حالات أورام حميدة سريعة النمو.[32] تكون بعض الأورام الخبيثة في الغالب غير منتشرة كما في حالة سرطان الخلايا القاعدية.[33] يمكن أن تكون الأشعة المقطعية والتصوير الشعاعي للصدر اختبارًا تشخيصيًا مفيدًا في تصور ورم حميد وتمييزه عن ورم خبيث. كلما كان الورم أصغر في الصورة الشعاعية، زادت احتمالية أن يكون حميدًا حيث أن 80% من عقيدات الرئة أقل من 2 قطرها سم حميدة. معظم العقيدات الحميدة عبارة عن كثافات ظليلة ملساء مع هوامش واضحة ولكن هذه ليست علامات حصرية للأورام الحميدة.[34]
التسرطن متعدد المراحل
تتكون الأورام من التسرطن، وهي عملية تؤدي فيها التغيرات الخلوية إلى تكوين السرطان. يتضمن التسرطن متعدد المراحل التغييرات الجينية أو اللاجينية المتتابعة على الحمض النووي للخلية، حيث تنتج كل خطوة ورمًا أكثر تقدمًا. غالبًا ما يتم تقسيمها إلى ثلاث مراحل؛ قد تحدث العديد من الطفرات في كل مرحلة. البدء هو مكان حدوث الطفرة الجينية الأولى في الخلية. التعزيز هو التوسع النسيلي (الانقسام المتكرر) لهذه الخلية المحولة إلى ورم مرئي يكون عادة حميدًا. بعد الترقية، قد يحدث تقدم حيث يتم اكتساب المزيد من الطفرات الجينية في مجموعة فرعية من الخلايا السرطانية. يؤدي التقدم إلى تغيير الورم الحميد إلى ورم خبيث.[4][35] من الأمثلة البارزة والمدروسة لهذه الظاهرة الورم الحميد الأنبوبي، وهو نوع شائع من ورم القولون الذي يعتبر مقدمة مهمة لسرطان القولون. تظهر الخلايا الموجودة في الأورام الغدية الأنبوبية، مثل معظم الأورام التي تتطور بشكل متكرر إلى سرطان، بعض التشوهات في نضج الخلايا وظهورها المعروفة باسم خلل التنسج. لا تظهر هذه التشوهات الخلوية في الأورام الحميدة التي نادرًا ما تتحول إلى سرطانية أو لا تتحول أبدًا إلى سرطانية، ولكنها تظهر في تشوهات الأنسجة الأخرى قبل السرطانية والتي لا تشكل كتلًا منفصلة، مثل الآفات السابقة للسرطان في عنق الرحم.
التشخيص
التصنيف
| الخلية الأصلية | نوع الخلية | الورم |
|---|---|---|
| الأديم الباطن | صفراوية | ورم الأقنية الصفراوية |
| قولونية | سليلة قولونية مستقيمية | |
| غدية | ورم غدي | |
| ورم حليمي | ||
| ورم غدي كيسي | ||
| كبدية | الورم الغدي ذو الخلايا الكبدية | |
| مشيمية | الرحى العدارية | |
| كلوية | ورم غدي أنبوبي كلوي | |
| طلائية | الورم الحليمي ذو الخلايا الحرشفية | |
| معدية | زوائد غدد قاع المعدة | |
| اللحمة المتوسطة | وعائية دموية | ورم وعائي دموي |
| عظمية | ورم عظمي | |
| غضروفية | ورم غضروفي | |
| نسيج دهني | ورم شحمي | |
| نسيج ضام | ورم ليفي | |
| وعاء لمفي | ورم وعائي لمفي | |
| عضلة ملساء | ورم عضلي أملس | |
| عضلة مخططة | ورم عضلي مخطط | |
| الأديم الظاهر | خلية دبقية | ورم نجمي |
| خلايا ميلانينية | وحمة | |
| سحايا | ورم سحائي | |
| عصبونات | ورم عصبي عقدي | |
| المراجع[36] | ||
غالباً وليس دائما تكون الأورام الحميدة مؤلفة من خلايا تحمل شبهاً قوياً للخلايا الطبيعية في جهازها الأصلي. تسمى هذه الأورام تبعاً للخلية أو النسيج الذي نشأت منه متبوعة باللاحقة "-oma" (بإستثناء السرطانة والساركوما، والتي تصنف عموماً ضمن السرطانات). على سبيل المثال الورم الشحمي (بالإنجليزية: Lipoma) هو ورم حميد شائع يتكون من خلايا دهنية (Lipocytes).
ورم غضروفي هو ورم حميد من الخلايا المكونة للغضروف (غضروفية). أورام هي أورام حميدة من الخلايا المكونة للغدة، وعادة ما يتم تحديد مزيد من زنزاناتهم أو الجهاز المنشأ، كما هو الحال في الورم الحميد الكبدي (ورم حميد من خلايا الكبد، أو الكبد الخلايا). تحتوي التراتومة على العديد من أنواع الخلايا مثل الجلد والعصب والدماغ والغدة الدرقية، من بين أمور أخرى، لأنها مشتقة من الخلايا الجرثومية [33] الورم العضلي عبارة عن مجموعة من الأورام الحميدة التي لها تمايز خلوي طبيعي نسبيًا ولكن بنية النسيج غير منظمة.[21] هناك عدد قليل من السرطانات ذات الأسماء «الحميدة» التي تم الاحتفاظ بها لأسباب تاريخية، بما في ذلك الورم الميلانيني (سرطان خلايا الجلد المصطبغة، أو الخلايا الصباغية) والورم المنوي (سرطان الخلايا التناسلية الذكرية).[37] غالبًا ما يُشار إلى علامات الجلد، وسلائل الحبال الصوتية، والأورام الحميدة المفرطة التنسج في القولون على أنها حميدة ولكنها في الواقع عبارة عن فرط نمو الأنسجة الطبيعية بدلاً من الأورام.[33]
العلاج
بعض الأورام الحميدة لا تحتاج إلى علاج. يمكن إزالة الآخرين إذا تسببوا في مشاكل مثل النوبات أو عدم الراحة أو مخاوف التجميل. عادة ما تكون الجراحة هي الطريقة الأكثر فعالية وتستخدم لعلاج معظم الأورام الحميدة. في بعض الحالات قد تكون العلاجات الأخرى مفيدة. يمكن علاج الأورام الغدية في المستقيم بالعلاج بالتصليب، وهو علاج يتم فيه استخدام المواد الكيميائية لتقليص الأوعية الدموية من أجل قطع إمدادات الدم.[38] لا تستجيب معظم الأورام الحميدة للعلاج الكيميائي أو العلاج الإشعاعي، على الرغم من وجود استثناءات؛ يتم علاج الأورام الحميدة بين الجمجمة أحيانًا بالعلاج الإشعاعي والعلاج الكيميائي في ظل ظروف معينة.[39][40] يمكن أيضًا استخدام الإشعاع لعلاج الأورام الوعائية في المستقيم.[38] وعادة ما تكون مقطوعة جراحيا أورام الجلد الحميدة ولكن علاجات أخرى مثل العلاج بالتبريد، كحت، تجفيف كهربي، العلاج بالليزر، جلدي، التقشير الكيميائي والأدوية الموضعية تستخدم.[41][42]
مراجع
- Clark WH (أكتوبر 1991)، "Tumour progression and the nature of cancer"، Br. J. Cancer، 64 (4): 631–44، doi:10.1038/bjc.1991.375، PMC 1977704، PMID 1911211.
- "PTEN hamartoma tumor syndromes"، Eur. J. Hum. Genet.، 16 (11): 1289–300، نوفمبر 2008، doi:10.1038/ejhg.2008.162، PMID 18781191.
- Brada M (فبراير 2013)، "Radiotherapy for benign brain tumours coming of age; example of vestibular schwannoma"، Radiother Oncol، 106 (2): 157–60، doi:10.1016/j.radonc.2013.01.009، PMID 23462704.
- Clark WH (أكتوبر 1991)، "Tumour progression and the nature of cancer"، Br. J. Cancer، 64 (4): 631–44، doi:10.1038/bjc.1991.375، PMID 1911211.
- Ober, William B.؛ Martini, Frederic (2006)، Fundamentals of anatomy & physiology، San Francisco: Pearson Benjamin Cummings، ISBN 0-321-31198-1.
- Wilson, Kathleen Atkins؛ Waugh, Anne؛ Chambers, Graeme؛ Grant, Allison؛ Ross, Janet (2006)، Ross and Wilson anatomy and physiology in health and illness، Edinburgh: Churchill Livingstone، ص. 53–54، ISBN 0-443-10101-9.
- "Tumours producing hypoglycaemia"، Diabetes Metab Rev، 7 (2): 79–91، يونيو 1991، doi:10.1002/dmr.5610070202، PMID 1665409.
- Grant CS (أكتوبر 2005)، "Insulinoma"، Best Pract Res Clin Gastroenterol، 19 (5): 783–98، doi:10.1016/j.bpg.2005.05.008، PMID 16253900.
- Charis Eng؛ DeLellis, Ronald A.؛ Lloyd, Ricardo V.؛ Phillipp U. Heitz (2004)، Pathology and genetics of tumours of endocrine organs، Lyon: IARC Press، ISBN 92-832-2416-7.
- "Small intestinal neoplasms"، J. Clin. Gastroenterol.، 33 (4): 267–82، أكتوبر 2001، doi:10.1097/00004836-200110000-00004، PMID 11588539.
- "Congenital melanocytic nevi needing treatment"، Dermatol Ther، 18 (2): 136–50، 2005، doi:10.1111/j.1529-8019.2005.05012.x، PMID 15953143.
- M. Zuber؛ F. Harder (2001)، Benign tumors of the colon and rectum، Munich: Zuckschwerdt: Surgical Treatment: Evidence-Based and Problem-Oriented.، مؤرشف من الأصل في 17 ديسمبر 2019.
- "PTEN hamartoma tumor syndrome: an overview"، Genet. Med.، 11 (10): 687–94، أكتوبر 2009، doi:10.1097/GIM.0b013e3181ac9aea، PMID 19668082.
- "Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome"، J. Med. Genet.، 41 (5): 323–6، مايو 2004، doi:10.1136/jmg.2004.018036، PMID 15121767.
- Eng C (نوفمبر 2000)، "Will the real Cowden syndrome please stand up: revised diagnostic criteria"، J. Med. Genet.، 37 (11): 828–30، doi:10.1136/jmg.37.11.828، PMID 11073535.
- Eng C (سبتمبر 2003)، "PTEN: one gene, many syndromes"، Hum. Mutat.، 22 (3): 183–98، doi:10.1002/humu.10257، PMID 12938083.
- "PTEN hamartoma tumor syndromes"، Eur. J. Hum. Genet.، 16 (11): 1289–300، نوفمبر 2008، doi:10.1038/ejhg.2008.162، PMID 18781191.
- Cohen MM (أغسطس 2005)، "Proteus syndrome: an update"، Am J Med Genet C Semin Med Genet، 137C (1): 38–52، doi:10.1002/ajmg.c.30063، PMID 16010681.
- "Familial adenomatous polyposis"، Am. J. Gastroenterol.، 101 (2): 385–98، فبراير 2006، PMID 16454848.
- "Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene"، J. Cell Sci.، 120 (Pt 19): 3327–35، أكتوبر 2007، doi:10.1242/jcs.03485، PMID 17881494.
- "Dysregulation of the TSC-mTOR pathway in human disease"، Nat. Genet.، 37 (1): 19–24، يناير 2005، doi:10.1038/ng1494، PMID 15624019.
- "The tuberous sclerosis complex"، N. Engl. J. Med.، 355 (13): 1345–56، سبتمبر 2006، doi:10.1056/NEJMra055323، PMID 17005952.
- Kwiatkowski DJ (يناير 2003)، "Tuberous sclerosis: from tubers to mTOR"، Ann. Hum. Genet.، 67 (Pt 1): 87–96، doi:10.1046/j.1469-1809.2003.00012.x، PMID 12556239.
- Maher ER (ديسمبر 2004)، "Von Hippel-Lindau disease"، Curr. Mol. Med.، 4 (8): 833–42، doi:10.2174/1566524043359827، PMID 15579030.
- Catherine D. Bachur, BA and Anne M. Comi, MD (2013)، Sturge-Weber Syndrome، Curr Treat Options Neurol. 2013 Oct; 15(5): 607–617.doi: 10.1007/s11940-013-0253-6،
{{استشهاد}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Kevin P. Boyd, MD, Bruce R. Korf, MD, PhD, and Amy Theos, MD (2009)، Neurofibromatosis type 1، J Am Acad Dermatol. 2009 Jul; 61(1): 1–16.doi: 10.1016/j.jaad.2008.12.051،
{{استشهاد}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Ashok R Asthagiri, MD, Dilys M Parry, PhD, John A Butman, MD, H Jeffrey Kim, MD, Ekaterini T Tsilou, MD, Prof. Zhengping Zhuang, MD, and Prof. Russell R Lonser, MD (2009)، Neurofibromatosis type 2، Lancet. 2009 Jun 6; 373(9679): 1974–1986. doi: 10.1016/S0140-6736(09)60259-2،
{{استشهاد}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Wilson, Kathleen Atkins؛ Waugh, Anne؛ Chambers, Graeme؛ Grant, Allison؛ Ross, Janet (2006)، Ross and Wilson anatomy and physiology in health and illness، Edinburgh: Churchill Livingstone، ص. 53–54، ISBN 0-443-10101-9.
- Nunn, Laura Silverstein؛ Silverstein, Alvin؛ Silverstein, Virginia B. (2006)، Cancer، Brookfield, Conn: Twenty-First Century Books، ص. 11–12، ISBN 0-7613-2833-5، مؤرشف من الأصل في 18 فبراير 2022.
- "Clinical and morphological features of undifferentiated monomorphous GH/TSH-secreting pituitary adenoma"، Eur. J. Endocrinol.، 140 (6): 528–37، يونيو 1999، doi:10.1530/eje.0.1400528، PMID 10366409.
- "Uncommon metastases from differentiated thyroid carcinoma" (PDF)، Hellenic Journal of Nuclear Medicine، 15 (3): 233–40، 2012، doi:10.1967/s002449910059 (غير نشط 1 سبتمبر 2020)، PMID 23106056، مؤرشف من الأصل (PDF) في 10 أغسطس 2017.
{{استشهاد بدورية محكمة}}: صيانة CS1: وصلة دوي غير نشطة منذ 2020 (link) - "Hamartoma: on occasion a rapidly growing tumor of the lung"، Radiology، 91 (5): 971–2، نوفمبر 1968، doi:10.1148/91.5.971، PMID 5681331.
- David Lowell Strayer؛ Raphael Rubin؛ Rubin, Emanuel (2008)، Rubin's pathology: clinicopathologic foundations of medicine، Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins، ص. 138–139، ISBN 978-0-7817-9516-6.
- Erasmus, J.J؛ Connolly, J.E؛ McAdams, H.P؛ Roggli, V.L. (2000)، "Solitary pulmonary nodules: Part I. Morphologic evaluation for differentiation of benign and malignant lesions"، Radiographics، 20 (1): 43–58، doi:10.1148/radiographics.20.1.g00ja0343، PMID 10682770.
- Barrett JC (أبريل 1993)، "Mechanisms of multistep carcinogenesis and carcinogen risk assessment"، Environ. Health Perspect.، 100: 9–20، doi:10.1289/ehp.931009، PMID 8354184.
- Wujcik, Debra؛ Yarbro, Connie Henke؛ Barbara H. Gobel (2011)، Cancer nursing: principles and practice، Boston: Jones and Bartlett Publishers، ISBN 0-7637-6357-8.
- Ramzi Cotran؛ Vinay Kumar؛ Tucker Collins (1999)، Robbins Pathologic Basis of Disease (ط. 6th)، W.B. Saunders، ISBN 0-7216-7335-X.
- M. Zuber؛ F. Harder (2001)، Benign tumors of the colon and rectum، Munich: Zuckschwerdt: Surgical Treatment: Evidence-Based and Problem-Oriented.، مؤرشف من الأصل في 8 يناير 2021.
- Brada M (فبراير 2013)، "Radiotherapy for benign brain tumours coming of age; example of vestibular schwannoma"، Radiother Oncol، 106 (2): 157–60، doi:10.1016/j.radonc.2013.01.009، PMID 23462704.
- "Chemotherapy, hormonal therapy, and immunotherapy for recurrent meningiomas"، J. Neurooncol.، 92 (1): 1–6، مارس 2009، doi:10.1007/s11060-008-9734-y، PMID 19023520.
- "Common benign skin tumors"، Am Fam Physician، 67 (4): 729–38، فبراير 2003، PMID 12613727.
- "Congenital melanocytic nevi: treatment modalities and management options"، Semin Cutan Med Surg، 26 (4): 231–40، ديسمبر 2007، doi:10.1016/j.sder.2008.03.007، PMID 18395671.
 بوابة طب
بوابة طب