Amide
In organic chemistry, an amide,[1][2][3] also known as an organic amide or a carboxamide, is a compound with the general formula R−C(=O)−NR′R″, where R, R', and R″ represent any group, typically organyl groups or hydrogen atoms.[4][5] The amide group is called a peptide bond when it is part of the main chain of a protein, and an isopeptide bond when it occurs in a side chain, such as in the amino acids asparagine and glutamine. It can be viewed as a derivative of a carboxylic acid (R−C(=O)−OH) with the hydroxyl group (−OH) replaced by an amine group (−NR′R″); or, equivalently, an acyl (alkanoyl) group (R−C(=O)−) joined to an amine group.
.svg.png.webp)
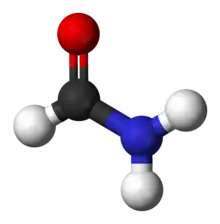

Common of amides are formamide (H−C(=O)−NH2), acetamide (H3C−C(=O)−NH2), benzamide (C6H5−C(=O)−NH2), and dimethylformamide (H−C(=O)−N(−CH3)2). Some uncommon examples of amides are N-chloroacetamide (H3C−C(=O)−NH−Cl) and chloroformamide (Cl−C(=O)−NH2).
Amides are qualified as primary, secondary, and tertiary according to whether the amine subgroup has the form −NH2, −NHR, or −NRR', where R and R' are groups other than hydrogen.
Nomenclature
The core −C(=O)−(N) of amides is called the amide group (specifically, carboxamide group).
In the usual nomenclature, one adds the term "amide" to the stem of the parent acid's name. For instance, the amide derived from acetic acid is named acetamide (CH3CONH2). IUPAC recommends ethanamide, but this and related formal names are rarely encountered. When the amide is derived from a primary or secondary amine, the substituents on nitrogen are indicated first in the name. Thus, the amide formed from dimethylamine and acetic acid is N,N-dimethylacetamide (CH3CONMe2, where Me = CH3). Usually even this name is simplified to dimethylacetamide. Cyclic amides are called lactams; they are necessarily secondary or tertiary amides.[6]
Applications
Amides are pervasive in nature and technology. Proteins and important plastics like Nylons, Aramid, Twaron, and Kevlar are polymers whose units are connected by amide groups (polyamides); these linkages are easily formed, confer structural rigidity, and resist hydrolysis. Amides include many other important biological compounds, as well as many drugs like paracetamol, penicillin and LSD.[7] Low-molecular-weight amides, such as dimethylformamide, are common solvents.
Structure and bonding
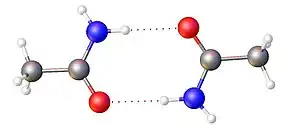
The lone pair of electrons on the nitrogen atom is delocalized into the carbonyl group, thus forming a partial double bond between nitrogen and carbon. In fact the O, C and N atoms have molecular orbitals occupied by delocalized electrons, forming a conjugated system. Consequently, the three bonds of the nitrogen in amides is not pyramidal (as in the amines) but planar. This planar restriction prevents rotations about the N linkage and thus has important consequences for the mechanical properties of bulk material of such molecules, and also for the configurational properties of macromolecules built by such bonds. The inability to rotate distinguishes amide groups from ester groups which allow rotation and thus create more flexible bulk material.
The C-C(O)NR2 core of amides is planar. The C=O distance is shorter than the C-N distance by almost 10%. The structure of an amide can be described also as a resonance between two alternative structures: neutral (A) and zwitterionic (B).
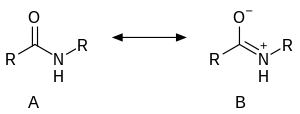
It is estimated that for acetamide, structure A makes a 62% contribution to the structure, while structure B makes a 28% contribution. (These figures do not sum to 100% because there are additional less-important resonance forms that are not depicted above). There is also a hydrogen bond present between the active groups hydrogen and nitrogen atoms.[9] Resonance is largely prevented in the very strained quinuclidone.
In their IR spectra, amides exhibit a moderately intense νCO band near 1650 cm−1. The energy of this band is about 60 cm-1 lower than for the νCO of esters and ketones. This difference reflects the contribution of the zwitterionic resonance structure.
Basicity
Compared to amines, amides are very weak bases. While the conjugate acid of an amine has a pKa of about 9.5, the conjugate acid of an amide has a pKa around −0.5. Therefore, amides do not have as clearly noticeable acid–base properties in water. This relative lack of basicity is explained by the withdrawing of electrons from the amine by the carbonyl. On the other hand, amides are much stronger bases than carboxylic acids, esters, aldehydes, and ketones (their conjugate acids' pKas are between −6 and −10).
The proton of a primary or secondary amide does not dissociate readily; its pKa is usually well above 15. Conversely, under extremely acidic conditions, the carbonyl oxygen can become protonated with a pKa of roughly −1. It is not only because of the positive charge on the nitrogen, but also because of the negative charge on the oxygen gained through resonance.
Hydrogen bonding and solubility
Because of the greater electronegativity of oxygen, the carbonyl (C=O) is a stronger dipole than the N–C dipole. The presence of a C=O dipole and, to a lesser extent a N–C dipole, allows amides to act as H-bond acceptors. In primary and secondary amides, the presence of N–H dipoles allows amides to function as H-bond donors as well. Thus amides can participate in hydrogen bonding with water and other protic solvents; the oxygen atom can accept hydrogen bonds from water and the N–H hydrogen atoms can donate H-bonds. As a result of interactions such as these, the water solubility of amides is greater than that of corresponding hydrocarbons. These hydrogen bonds are also have an important role in the secondary structure of proteins.
The solubilities of amides and esters are roughly comparable. Typically amides are less soluble than comparable amines and carboxylic acids since these compounds can both donate and accept hydrogen bonds. Tertiary amides, with the important exception of N,N-dimethylformamide, exhibit low solubility in water.
Reactions
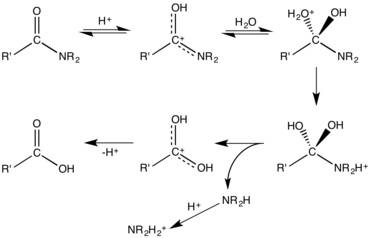 Mechanism for acid-mediated hydrolysis of an amide.[10]
Mechanism for acid-mediated hydrolysis of an amide.[10]
Amides undergo many chemical reactions, although they are less reactive than esters. Amides hydrolyse in hot alkali as well as in strong acidic conditions. Acidic conditions yield the carboxylic acid and the ammonium ion while basic hydrolysis yield the carboxylate ion and ammonia. The protonation of the initially generated amine under acidic conditions and the deprotonation of the initially generated carboxylic acid under basic conditions render these processes non-catalytic and irreversible. Amides are also versatile precursors to many other functional groups. Electrophiles react with the carbonyl oxygen. This step often precedes hydrolysis, which is catalyzed by both Brønsted acids and Lewis acids. Enzymes, e.g. peptidases and artificial catalysts, are known to accelerate the hydrolysis reactions.
| Reaction name | Product | Comment |
|---|---|---|
| Dehydration | Nitrile | Reagent: phosphorus pentoxide; benzenesulfonyl chloride; TFAA/py[11] |
| Hofmann rearrangement | Amine with one fewer carbon atom | Reagents: bromine and sodium hydroxide |
| Amide reduction | Amine | Reagent: lithium aluminium hydride followed by hydrolysis |
| Vilsmeier–Haack reaction | Aldehyde (via imine) | POCl3, aromatic substrate, formamide |
| Bischler–Napieralski reaction | Cyclic imine | POCl3, SOCl2, etc. |
Synthesis
From carboxylic acids and related compounds
Amides are usually prepared by coupling carboxylic acid with an amine. The direct reaction generally requires high temperatures to drive off the water:
- RCO2H + R'2NH → RCO−2 + R'2NH+2
- RCO−2 + R'2NH2 → RC(O)NR'2 + H2O
A set of common methods involve "activating" the carboxylic acid by converting it to a better electrophile. Thus, esters, acid chlorides (Schotten-Baumann reaction), or anhydrides (Lumière–Barbier method) all react with amines to give amides:
- RCO2R" + R'2NH → RC(O)NR'2 + R"OH
- RCOCl + 2R'2NH → RC(O)NR'2 + R'2NH+2Cl−
- (RCO)2O + R'2NH → RC(O)NR'2 + RCO2H
Peptide synthesis use coupling agents such as HATU, HOBt, or PyBOP.[12]
From nitriles
The hydrolysis of nitriles is conducted on an industrial scale to produce fatty amines.[13] Laboratory procedures are also available.[14]
Specialty routes
Many specialized methods also yield amides.[15] A variety of reagents, e.g. tris(2,2,2-trifluoroethyl) borate have been developed for specialized applications.[16][17]
| Reaction name | Substrate | Details |
|---|---|---|
| Beckmann rearrangement | Cyclic ketone | Reagent: hydroxylamine and acid |
| Schmidt reaction | Ketones | Reagent: hydrazoic acid |
| Willgerodt–Kindler reaction | Aryl alkyl ketones | Sulfur and morpholine |
| Passerini reaction | Carboxylic acid, ketone or aldehyde | |
| Ugi reaction | Isocyanide, carboxylic acid, ketone, primary amine | |
| Bodroux reaction[18][19] | Carboxylic acid, Grignard reagent with an aniline derivative ArNHR' | 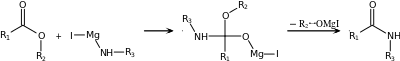 |
| Chapman rearrangement[20][21] | Aryl imino ether | For N,N-diaryl amides. The reaction mechanism is based on a nucleophilic aromatic substitution.[22] |
| Leuckart amide synthesis[23] | Isocyanate | Reaction of arene with isocyanate catalysed by aluminium trichloride, formation of aromatic amide. |
| Ritter reaction[24] | Alkenes, alcohols, or other carbonium ion sources | Secondary amides via an addition reaction between a nitrile and a carbonium ion in the presence of concentrated acids. |
| Photolytic addition of formamide to olefins[25] | Terminal alkenes | A free radical homologation reaction between a terminal alkene and formamide. |
| Ester aminolysis[26][27][28] | Esters | Base catalyzed reaction of esters with various amines to form alcohols and amides. |
| Dehydrogenative coupling[29] | alcohol, amine | requires ruthenium dehydrogenation catalyst |
| Transamidation[30][31] | amide | typically slow |
| Amine α-oxidation[32] | alkyl amine | requires gold catalysts |
References
- "Amide definition and meaning - Collins English Dictionary". www.collinsdictionary.com. Retrieved 15 April 2018.
- "amide". The American Heritage Dictionary of the English Language (5th ed.). HarperCollins.
- "amide - Definition of amide in English by Oxford Dictionaries". Oxford Dictionaries – English. Archived from the original on 2 April 2015. Retrieved 15 April 2018.
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "amides". doi:10.1351/goldbook.A00266
- "Chapter 21: Amides and Imides". Nomenclature of Organic Compounds. 1974. pp. 166–173. doi:10.1021/ba-1974-0126.ch021. ISBN 9780841201910.
{{cite book}}:|work=ignored (help) - Organic Chemistry IUPAC Nomenclature. Rules C-821. Amides http://www.acdlabs.com/iupac/nomenclature/79/r79_540.htm Archived 21 January 2011 at the Wayback Machine
- Boonen, Jente; Bronselaer, Antoon; Nielandt, Joachim; Veryser, Lieselotte; De Tré, Guy; De Spiegeleer, Bart (2012). "Alkamid database: Chemistry, occurrence and functionality of plant N-alkylamides" (PDF). Journal of Ethnopharmacology. 142 (3): 563–590. doi:10.1016/j.jep.2012.05.038. hdl:1854/LU-2133714. PMID 22659196. Archived (PDF) from the original on 9 October 2022.
- Bats, Jan W.; Haberecht, Monika C.; Wagner, Matthias (2003). "A new refinement of the orthorhombic polymorph of acetamide". Acta Crystallographica Section E. 59 (10): o1483–o1485. doi:10.1107/S1600536803019494.
- Kemnitz, Carl R.; Loewen, Mark J. (2007). ""Amide Resonance" Correlates with a Breadth of C−N Rotation Barriers". Journal of the American Chemical Society. 129 (9): 2521–8. doi:10.1021/ja0663024. PMID 17295481.
- Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, ISBN 978-0-471-72091-1
- U.S. Patent 5,935,953
- Valeur, Eric; Bradley, Mark (2009). "Amide bond formation: beyond the myth of coupling reagents". Chem. Soc. Rev. 38 (2): 606–631. doi:10.1039/B701677H. PMID 19169468. S2CID 14950926.
- Eller, Karsten; Henkes, Erhard; Rossbacher, Roland; Höke, Hartmut (2000). "Amines, Aliphatic". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a02_001.
- Wenner, Wilhelm (1952). "Phenylacetamide". Organic Syntheses. 32: 92. doi:10.15227/orgsyn.032.0092.
- Montalbetti, Christian A. G. N.; Falque, Virginie (14 November 2005). "Amide bond formation and peptide coupling". Tetrahedron. 61 (46): 10827–10852. doi:10.1016/j.tet.2005.08.031.
- "Tris(2,2,2-trifluoroethyl) borate 97% | Sigma-Aldrich". www.sigmaaldrich.com. Retrieved 22 September 2016.
- Sabatini, Marco T.; Boulton, Lee T.; Sheppard, Tom D. (1 September 2017). "Borate esters: Simple catalysts for the sustainable synthesis of complex amides". Science Advances. 3 (9): e1701028. Bibcode:2017SciA....3E1028S. doi:10.1126/sciadv.1701028. PMC 5609808. PMID 28948222.
- Bodroux F. (1905). Bull. Soc. Chim. France. 33: 831.
{{cite journal}}: CS1 maint: untitled periodical (link) - "Bodroux reaction". Institute of Chemistry, Skopje, Macedonia. Archived from the original on 24 September 2015. Retrieved 23 May 2007.
- Schulenberg, J. W.; Archer, S. (1965). The Chapman Rearrangement. pp. 1–51. doi:10.1002/0471264180.or014.01. ISBN 978-0471264187.
{{cite book}}:|journal=ignored (help) - Chapman, Arthur William (1925). "CCLXIX.—Imino-aryl ethers. Part III. The molecular rearrangement of N-phenylbenziminophenyl ether". Journal of the Chemical Society, Transactions. 127: 1992–1998. doi:10.1039/CT9252701992.
- March, Jerry (1966). Advanced organic Chemistry, Reactions, mechanisms and structure (3rd ed.). Wiley. ISBN 978-0-471-85472-2.
- Leuckart, R. (1885). "Ueber einige Reaktionen der aromatischen Cyanate". Berichte der deutschen chemischen Gesellschaft. 18: 873–877. doi:10.1002/cber.188501801182.
- Adams, Rodger; Krimen, L.I.; Cota, Donald J. (1969). Organic Reaction Volume 17. London: John Wiley & Sons, Inc. pp. 213–326. doi:10.1002/0471264180. ISBN 9780471196150.
- Monson, Richard (1971). Advanced Organic Synthesis: Methods and Techniques (PDF). New York: Academic Press. p. 141. ISBN 978-0124336803. Archived (PDF) from the original on 9 October 2022.
- Corson, B. B.; Scott, R. W.; Vose, C. E. (1941). "Cyanoacetamide". Organic Syntheses. 1: 179. doi:10.15227/orgsyn.009.0036.
- Jacobs, W. A. (1941). "Chloroacetamide". Organic Syntheses. 1: 153. doi:10.15227/orgsyn.007.0016.
- Kleinberg, J.; Audrieth, L. F. (1955). "Lactamide". Organic Syntheses. 3: 516. doi:10.15227/orgsyn.021.0071.
- Gunanathan, C.; Ben-David, Y.; Milstein, D. (2007). "Direct Synthesis of Amides from Alcohols and Amines with Liberation of H2". Science. 317 (5839): 790–2. Bibcode:2007Sci...317..790G. doi:10.1126/science.1145295. PMID 17690291. S2CID 43671648.
- T. A. Dineen; M. A. Zajac; A. G. Myers (2006). "Efficient Transamidation of Primary Carboxamides by in situ Activation with N,N-Dialkylformamide Dimethyl Acetals". J. Am. Chem. Soc. 128 (50): 16406–16409. doi:10.1021/ja066728i. PMID 17165798.
- Emma L. Baker; Michael M. Yamano; Yujing Zhou; Sarah M. Anthony; Neil K. Garg (2016). "A two-step approach to achieve secondary amide transamidation enabled by nickel catalysis". Nature Communications. 7: 11554. Bibcode:2016NatCo...711554B. doi:10.1038/ncomms11554. PMC 4876455. PMID 27199089.
- P. Chatterjee; H. Wang; J. S. Manzano; U. Kanbur; A. D. Sadow; I. I. Slowing (2022). "Surface ligands enhance the catalytic activity of supported Au nanoparticles for the aerobic α-oxidation of amines to amides". Catal. Sci. Technol. 12 (6): 1922–1933. doi:10.1039/D1CY02121D. S2CID 246575960.