Fatty-acid metabolism disorder
A broad classification for genetic disorders that result from an inability of the body to produce or utilize one enzyme that is required to oxidize fatty acids. The enzyme can be missing or improperly constructed, resulting in it not working. This leaves the body unable to produce energy within the liver and muscles from fatty acid sources.[1]
| Fatty-acid metabolism disorder | |
|---|---|
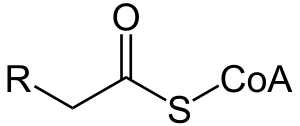 | |
| Acyl-CoA, one of the compounds involved in fatty acid metabolism | |
| Specialty | Endocrinology |
The body's primary source of energy is glucose; however, when all the glucose in the body has been expended, a normal body digests fats. Individuals with a fatty-acid metabolism disorder are unable to metabolize this fat source for energy, halting bodily processes.[1] Most individuals with a fatty-acid metabolism disorder are able to live a normal active life with simple adjustments to diet and medications.
If left undiagnosed many complications can arise. When in need of glucose the body of a person with a fatty-acid metabolism disorder will still send fats to the liver. The fats are broken down to fatty acids. The fatty acids are then transported to the target cells but are unable to be broken down, resulting in a build-up of fatty acids in the liver and other internal organs.
Fatty-acid metabolism disorders are sometimes classified with the lipid metabolism disorders,[2] but in other contexts they are considered a distinct category.
Types
Incomplete list of various fatty-acid metabolism disorders.[1]
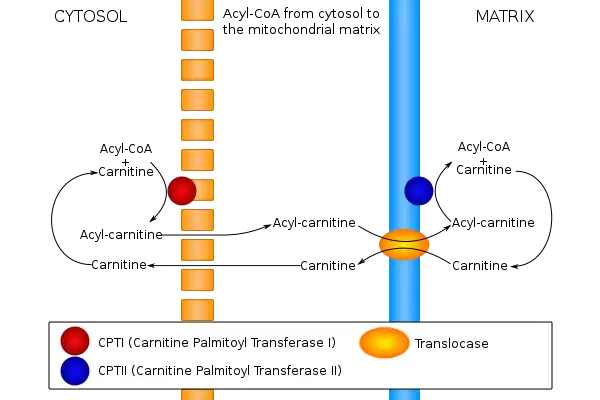
- Carnitine Transport Defect
- Carnitine-acylcarnitine translocase deficiency (CACT)
- Carnitine Palmitoyl Transferase I & II ( CPT I deficiency & CPT II deficiency)
- 2,4 Dienoyl-CoA Reductase Deficiency
- Electron Transfer Flavoprotein (ETF) Dehydrogenase Deficiency (GAII & MADD)
- 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency (HMG deficiency)
- Very long-chain acyl-coenzyme A dehydrogenase deficiency (VLCAD deficiency)
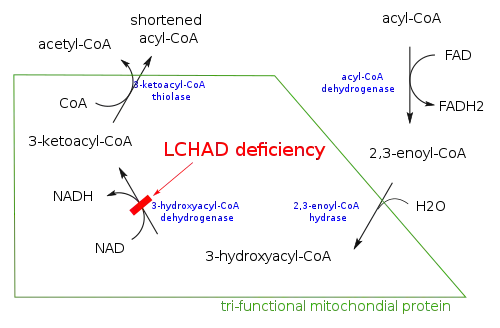
- Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency (LCHAD deficiency)
- Medium-chain acyl-coenzyme A dehydrogenase deficiency (MCAD deficiency)
- Short-chain acyl-coenzyme A dehydrogenase deficiency (SCAD deficiency)
- 3-hydroxyacyl-coenzyme A dehydrogenase deficiency (M/SCHAD deficiency)
Oxidation
The term fatty acid oxidation disorder (FAOD) is sometimes used, especially when there is an emphasis on the oxidation of the fatty acid.[3]
In addition to the fetal complications, they can also cause complications for the mother during pregnancy.[4]
Examples include:
Symptoms and signs
Causes
Fatty-acid metabolism disorders result when both parents of the diagnosed subject are carriers of a defective gene. This is known as an autosomal recessive disorder. Two parts of a recessive gene are required to activate the disease. If only one part of the gene is present then the individual is only a carrier and shows no symptoms of the disease. If both mutated genes are present, the individual will be symptomatic. Like most autosomal recessive disorders, when both parents are carriers, there is a 25% chance for each child to inherit the disease.[7]
Diagnosis
Diagnosis of Fatty-acid metabolism disorder requires extensive lab testing.
Normally, in cases of hypoglycaemia, triglycerides and fatty acids are metabolised to provide glucose/energy. However, in this process, ketones are also produced and ketotic hypoglycaemia is expected. However, in cases where fatty acid metabolism is impaired, a non-ketotic hypoglycaemia may be the result, due to a break in the metabolic pathways for fatty-acid metabolism.
Treatment
The primary treatment method for fatty-acid metabolism disorders is dietary modification. It is essential that the blood-glucose levels remain at adequate levels to prevent the body from moving fat to the liver for energy. This involves snacking on low-fat, high-carbohydrate nutrients every 2–6 hours. However, some adults and children can sleep for 8–10 hours through the night without snacking.[1][7]
Drugs
Carnitor - an L-carnitine supplement that has shown to improve the body's metabolism in individuals with low L-carnitine levels. It is only useful for Specific fatty-acid metabolism disease.[1]
References
- Gould D. (2011). "List of FODs and symptoms".
{{cite journal}}: Cite journal requires|journal=(help) - "Lipid Metabolism: Hereditary Metabolic Disorders: Merck Manual Home Edition". Retrieved 2009-03-11.
- Shekhawat PS, Matern D, Strauss AW (May 2005). "Fetal fatty acid oxidation disorders, their effect on maternal health and neonatal outcome: impact of expanded newborn screening on their diagnosis and management". Pediatr. Res. 57 (5 Pt 2): 78R–86R. doi:10.1203/01.PDR.0000159631.63843.3E. PMC 3582391. PMID 15817498.
- Ibdah JA, Bennett MJ, Rinaldo P, et al. (June 1999). "A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women". N. Engl. J. Med. 340 (22): 1723–31. doi:10.1056/NEJM199906033402204. PMID 10352164.
- "Fatty Acid and Glycerol Metabolism Disorders: Inherited Disorders of Metabolism: Merck Manual Professional". Retrieved 2009-03-11.
- Longo N, Amat di San Filippo C, Pasquali M (May 2006). "Disorders of carnitine transport and the carnitine cycle". Am J Med Genet C Semin Med Genet. 142C (2): 77–85. doi:10.1002/ajmg.c.30087. PMC 2557099. PMID 16602102.
- "Fatty Acid Oxidation Disorders" (PDF). Retrieved 2011-11-11.