Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency
Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency is a rare autosomal recessive fatty acid oxidation disorder[1] that prevents the body from converting certain fats into energy. This can become life-threatening, particularly during periods of fasting.
| Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency | |
|---|---|
| Other names | LCHAD deficiency |
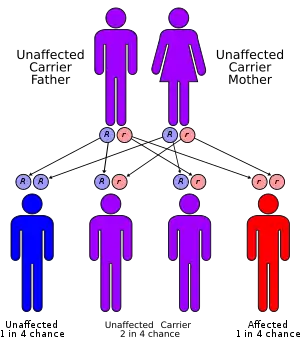 | |
| Long-chain 3-hydroxyacyl-coenzyme; A dehydrogenase deficiency has an autosomal recessive pattern of inheritance. | |
Symptoms and signs
Typically, initial signs and symptoms of this disorder occur during infancy or early childhood and can include feeding difficulties, lethargy, hypoglycemia, hypotonia, liver problems, and abnormalities in the retina. Muscle pain, a breakdown of muscle tissue, and abnormalities in the nervous system that affect arms and legs (peripheral neuropathy) may occur later in childhood. There is also a risk for complications such as life-threatening heart and breathing problems, coma, and sudden unexpected death. Episodes of LCHAD deficiency can be triggered by periods of fasting or by illnesses such as viral infections.
Genetics
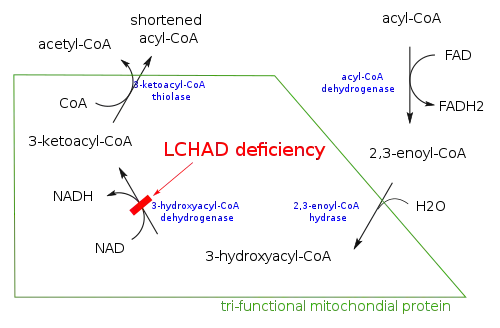
Mutations in the HADHA gene lead to inadequate levels of an enzyme called long-chain 3-hydroxyacyl-coenzyme A (CoA) dehydrogenase, which is part of a protein complex known as mitochondrial trifunctional protein. Long-chain fatty acids from food and body fat cannot be metabolized and processed without sufficient levels of this enzyme. As a result, these fatty acids are not converted to energy, which can lead to characteristic features of this disorder, such as lethargy and hypoglycemia. Long-chain fatty acids or partially metabolized fatty acids may build up in tissues and damage the liver, heart, retina, and muscles, causing more serious complications.
Prognosis
A 2001 study followed up on 50 patients. Of these 38% died in childhood while the rest suffered from problems with morbidity.[2]
References
- Reference, Genetics Home. "LCHAD deficiency". Genetics Home Reference. Retrieved 2017-02-27.
- Boer, Margarethe E. J. den; Wanders, Ronald J. A.; Morris, Andrew A. M.; IJlst, Lodewijk; Heymans, Hugo S. A.; Wijburg, Frits A. (2002-01-01). "Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency: Clinical Presentation and Follow-Up of 50 Patients". Pediatrics. 109 (1): 99–104. doi:10.1542/peds.109.1.99. ISSN 0031-4005. PMID 11773547.
External links