Malonyl-CoA decarboxylase deficiency
Malonyl-CoA decarboxylase deficiency (MCD) is an autosomal-recessive[1] metabolic disorder caused by a genetic mutation that disrupts the activity of Malonyl-CoA decarboxylase. This enzyme breaks down Malonyl-CoA (a fatty acid precursor and a fatty acid oxidation blocker) into acetyl-CoA and carbon dioxide.
| Malonyl-CoA decarboxylase deficiency | |
|---|---|
| Other names | Malonic aciduria |
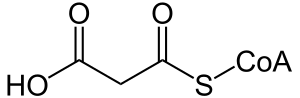 | |
| Malonyl-CoA | |
Signs and symptoms
The signs and symptoms of this disorder typically appear in early childhood. Almost all affected children have delayed development. Additional signs and symptoms can include weak muscle tone (hypotonia), seizures, diarrhea, vomiting, and low blood sugar (hypoglycemia). A heart condition called cardiomyopathy, which weakens and enlarges the heart muscle, is another common feature of malonyl-CoA decarboxylase deficiency.
Some common symptoms in Malonyl-CoA decarboxylase deficiency, such as cardiomyopathy and metabolic acidosis, are triggered by the high concentrations of Malonyl-CoA in the cytoplasm. High levels of Malonyl-CoA will inhibit β-oxidation of fatty acids through deactivating the carrier of fatty acyl group, CPT1, and thus, blocking fatty acids from going into the mitochondrial matrix for oxidation.[2]
A research conducted in Netherlands has suggested that carnitine supplements and a low fat diet may help to reduce the level of malonic acid in our body.[3]
Genetics

Malonyl-CoA decarboxylase deficiency is caused by mutations in the MLYCD gene, located on chromosome 16q24.[4] The gene encodes the enzyme malonyl-CoA decarboxylase. Within cells, this enzyme helps regulate the formation and breakdown of a certain group of fats called fatty acids.
Many tissues, including heart muscle, use fatty acids as a major source of energy. Mutations in the MLYCD gene reduce or eliminate the function of malonyl-CoA decarboxylase. A lack of this enzyme disrupts the normal balance of fatty acid formation and breakdown. As a result, fatty acids cannot be converted to energy, which can lead to characteristic features of this disorder, such as low blood sugar and cardiomyopathy. By-products of fatty acid processing build up in tissues, which also contributes to the signs and symptoms of malonyl-CoA decarboxylase deficiency.
Malonyl-CoA decarboxylase deficiency is inherited in an autosomal recessive pattern.[1] This means that the defective gene is located on an autosome (chromosome 16 is an autosome), and two copies of the defective gene - one inherited from each parent - are required to be born with the disorder. The parents of a child with an autosomal recessive disorder both carry one copy of the defective gene, but are usually not affected by the disorder.
Malonyl-CoA decarboxylase deficiency is extremely rare, evidence suggests that it is caused by the abnormality in the protein transcription regulation.[2] Looking at the molecular basis, two distinct homozygous mutations are found to cause Malonyl-CoA Decarboxylase deficiency in human. The first mutation is the transversion of gene from C to G causing a premature stop signal in the protein. The second mutation is a base pair insertion in the mature RNA that will eventually result in the protein truncation.[5]
A research has also confirmed that the homozygous mutation which eventually leads to MCD deficiency is caused by the isodisomy of maternal UPD. This indicates that such disease is likely to be inherited from mother’s gene profile, not from paternal source.[3]
Pathophysiology
Without the enzymatic activity of Malonyl-CoA decarboxylase, cellular Mal-CoA increases so dramatically that at the end it is instead broken down by an unspecific short-chain acyl-CoA hydrolase, which produces malonic acid and CoA. Malonic acid is a Krebs cycle inhibitor, preventing the cells to make ATP through oxidation. In this condition, the cells, to make ATP, are forced to increase glycolysis, which produces lactic acid as a by-product. The increase of lactic and malonic acid drastically lowers blood pH, and causes both lactic and malonic aciduria (acidic urine). This condition is very rare, as fewer than 20 cases have been reported.
By 1999, only seven cases of Malonyl- CoA decarboxylase deficiency had been reported in human in Australia; however, this deficiency predominately occurs during childhood. Patients from the seven reported cases of Malonyl- CoA decarboxylase deficiency have an age range between 4 days to 13 years, and they all have the common symptom of delayed neurological development.[5] Similar study was conducted in Netherland, and found seventeen reported cases of Malonyl- CoA decarboxylase deficiency in children age range from 8 days to 12 years.[2]
Although we have not yet gained a clear understanding of the pathogenic mechanism of this deficiency, some researchers have suggested a brain-specific interaction between Malonyl-CoA and CTP1 enzyme which may leads to unexplained symptoms of the MCD deficiency.[3]
Research has found that large amount of MCD are detached in the hypothalamus and cortex of the brain where high levels of lipogenic enzymes are found, indicating that MCD plays a role in lipid synthesis in the brain.[2] Disturbed interaction between Malonyl-CoA and CPT1 may also contributed to abnormal brain development.[2]
Malonyl-CoA decarboxylase plays an important role in the β-oxidation processes in both mitochondria and peroxisome.[5] Some other authors have also hypothesized that it is the MCD deficiency induced inhibition of peroxisomal β-oxidation that contributes to the development delay.[5]
See also
References
- MacPhee, G. B.; Logan, R. W.; Mitchell, J. S.; Howells, D. W.; Tsotsis, E.; Thorburn, D. R. (October 1993). "Malonyl coenzyme a decarboxylase deficiency". Archives of Disease in Childhood. 69 (4): 433–436. doi:10.1136/adc.69.4.433. PMC 1029550. PMID 8259873.
- de Wit MC, de Coo IF, Verbeek E, Schot R, Schoonderwoerd GC, Duran M, de Klerk JB, Huijmans JG, Lequin MH, Verheijen FW, Mancini GM (February 2006). "Brain abnormalities in a case of malonyl-CoA decarboxylase deficiency". Molecular Genetics and Metabolism. 87 (2): 102–6. doi:10.1016/j.ymgme.2005.09.009. PMID 16275149.
- Malvagia S, Papi L, Morrone A, Donati MA, Ciani F, Pasquini E, la Marca G, Scholte HR, Genuardi M, Zammarchi E (November 2007). "Fatal malonyl CoA decarboxylase deficiency due to maternal uniparental isodisomy of the telomeric end of chromosome 16". Annals of Human Genetics. 71 (Pt 6): 705–12. doi:10.1111/j.1469-1809.2007.00373.x. PMID 17535268. S2CID 35678278.
- Online Mendelian Inheritance in Man (OMIM): 606761
- FitzPatrick, DR; Hill, A; Tolmie, JL; Thorburn, DR; Christodoulou, J (August 1999). "The molecular basis of malonyl-CoA decarboxylase deficiency". American Journal of Human Genetics. 65 (2): 318–26. doi:10.1086/302492. PMC 1377930. PMID 10417274.
External links
- Malonyl-CoA decarboxylase deficiency at NLM Genetics Home Reference