Mitochondrial trifunctional protein deficiency
Mitochondrial trifunctional protein deficiency (MTP deficiency or MTPD) is an autosomal recessive fatty acid oxidation disorder[4] that prevents the body from converting certain fats to energy, particularly during periods without food.[5][6] People with this disorder have inadequate levels of an enzyme that breaks down a certain group of fats called long-chain fatty acids.[6]
| Mitochondrial trifunctional protein deficiency | |
|---|---|
| Other names | TFP deficiency[1] |
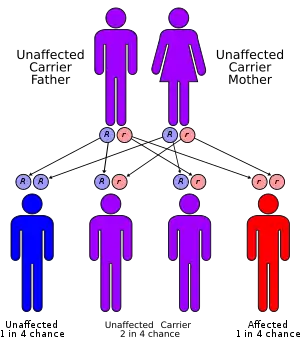 | |
| Mitochondrial trifunctional protein deficiency has an autosomal recessive pattern of inheritance | |
| Symptoms | Cardiomyopathy, skeletal myopathy [2] |
| Types | Mutations in the HADHA and HADHB gene[2] |
| Diagnostic method | CBC, Urine test[3] |
| Treatment | Low fat diet, Limited exercise[3] |
Signs and symptoms
The presentation of mitochondrial trifunctional protein deficiency may begin during infancy, features that occur are: low blood sugar, weak muscle tone, and liver problems. Infants with this disorder are at risk for heart problems, breathing difficulties, and pigmentary retinopathy. Signs and symptoms of mitochondrial trifunctional protein deficiency that may begin after infancy include hypotonia, muscle pain, a breakdown of muscle tissue, and a loss of sensation in the extremities called peripheral neuropathy. Some who have MTP deficiency show a progressive course associated with myopathy, and recurrent rhabdomyolysis.[2][6][7]
Genetics
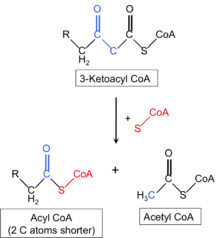
The genetics of mitochondrial trifunctional protein deficiency is based on mutations in the HADHA[8] and HADHB[9] genes which cause this disorder. These genes each provide instructions for making part of an enzyme complex called mitochondrial trifunctional protein. This enzyme complex functions in mitochondria, the energy-producing centers within cells: mitochondrial trifunctional protein contains three enzymes that each perform a different function. This enzyme complex is required to metabolize a group of fats called long-chain fatty acids. These fatty acids are stored in the body's fat tissues and are a major source of energy for the heart and muscles. During periods of fasting, fatty acids are also an important energy source for the liver and other tissues.[10][11][12]
Mutations in the HADHA or HADHB genes that cause mitochondrial trifunctional protein deficiency disrupt all functions of this enzyme complex.[13] Without enough of this enzyme complex, long-chain fatty acids cannot be metabolized. As a result, these fatty acids are not converted to energy, which can lead to some features of this disorder. Long-chain fatty acids may also build up and damage the liver, heart, and muscles. This abnormal buildup causes other symptoms of mitochondrial trifunctional protein deficiency.
The mechanism of this condition indicates that the mitochondrial trifunction protein catalyzes 3 steps in mitochondrial beta-oxidation of fatty acids: long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), long-chain enoyl-CoA hydratase, and long-chain thiolase activities. Trifunctional protein deficiency is characterized by decreased activity of all 3 enzymes. Clinically, trifunctional protein deficiency usually results in sudden unexplained infant death, cardiomyopathy, or skeletal myopathy.[14][11][12]
Diagnosis
Diagnosis of mitochondrial trifunctional protein deficiency is often confirmed using tandem mass spectrometry.[4] Genetic counseling is available for this condition. Additionally the following exams are available:
Treatment
Management for mitochondrial trifunctional protein deficiency entails the following:[7]
- Avoiding factors that might precipitate condition
- Glucose
- Low fat/high carbohydrate nutrition
See also
- Long-chain acyl-CoA dehydrogenase
References
- "Mitochondrial trifunctional protein deficiency | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 31 July 2019.
- "Mitochondrial trifunctional protein deficiency | Genetic and Rare Diseases Information Center(GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 12 November 2016.
- RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: Mitochondrial trifunctional protein deficiency". www.orpha.net. Retrieved 2016-11-12.
- Solish JO, Singh RH (2002). "Management of fatty acid oxidation disorders: a survey of current treatment strategies". J Am Diet Assoc. 102 (12): 1800–1803. doi:10.1016/S0002-8223(02)90386-X. PMID 12487544.subscription needed
- "OMIM Entry - # 609015 - MITOCHONDRIAL TRIFUNCTIONAL PROTEIN DEFICIENCY; MTPD". omim.org. Retrieved 2016-11-05.
- Reference, Genetics Home. "mitochondrial trifunctional protein deficiency". Genetics Home Reference. Retrieved 2016-10-28.
- Swaiman, Kenneth F.; Ashwal, Stephen; Ferriero, Donna M.; Schor, Nina F. (2014). Swaiman's Pediatric Neurology: Principles and Practice. Elsevier Health Sciences. pp. 461, 1638. ISBN 978-0323089111. Retrieved 12 November 2016.
- Reference, Genetics Home. "HADHA gene". Genetics Home Reference. Retrieved 2016-11-05.
- Reference, Genetics Home. "HADHB gene". Genetics Home Reference. Retrieved 2016-11-05.
- "Long-Chain Acyl CoA Dehydrogenase Deficiency: Background, Pathophysiology, Epidemiology". eMedicine. 24 March 2016. Retrieved 12 November 2016.
- "HADHA hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase (trifunctional protein), alpha subunit [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov. Retrieved 12 November 2016.
- "Home - Gene - NCBI". www.ncbi.nlm.nih.gov. Retrieved 12 November 2016.
- "OMIM Entry - * 600890 - HYDROXYACYL-CoA DEHYDROGENASE/3-KETOACYL-CoA THIOLASE/ENOYL-CoA HYDRATASE, ALPHA SUBUNIT; HADHA". omim.org. Retrieved 5 November 2016.
- Rector, R. Scott; Payne, R. Mark; Ibdah, Jamal A. (1 January 2008). "Mitochondrial Trifunctional Protein Defects: Clinical Implications and Therapeutic Approaches". Adv Drug Deliv Rev. 60 (13–14): 1488–1496. doi:10.1016/j.addr.2008.04.014. ISSN 0169-409X. PMC 2848452. PMID 18652860.
Further reading
- Nyhan, William L.; Hoffman, Georg F.; Barshop, Bruce A.; Al-Aqeel, Aida I. (2012). Atlas of Inherited Metabolic Diseases 3E. CRC Press. ISBN 9781444149487.
- Thöny, Beat; Duran, Marinus; Gibson, K. Michael; Dionisi-Vici, Carlo (2013). Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases. Springer. ISBN 9783642403378.
External links
 Media related to Mitochondrial trifunctional protein deficiency at Wikimedia Commons
Media related to Mitochondrial trifunctional protein deficiency at Wikimedia Commons