Multiple sclerosis diagnosis
Current standards for diagnosing multiple sclerosis (MS) are based on the 2018 revision of McDonald criteria. They rely on MRI detection (or clinical demonstration) of demyelinating lesions in the CNS, which are distributed in space (DIS) and in time (DIT). It is also a requirement that any possible known disease that produces demyelinating lesions is ruled out before applying McDonald's criteria.
| Diagnosis of multiple sclerosis | |
|---|---|
 Animation showing dissemination of brain lesions in time and space as demonstrated by monthly MRI studies along a year | |
| Purpose | diagnosis via lab test, imaging and symptoms |
This last requirement makes MS an ill-defined entity, whose borders change every time that a new disease is set apart. Some cases previously considered MS are now considered distinct conditions, like Neuromyelitis optica or antiMOG associated encephalomyelitis. Because of the requirement of distributed lesions, a single lesion (RIS) is not considered MS. For the same reason, the prodromal stage of MS (the unknown condition that causes the lesions) would not be considered as MS if it could be found.
Sometimes the diagnosis must be retrospective, relying on gradual worsening of neurological signs/symptoms, due to the lack of understanding of the pathogenicity driving disease progression.[1] However, the only definite diagnosis of MS is post-mortem autopsy, where lesions typical of MS can be detected through histopathological techniques.[2][3]
Overview
Multiple sclerosis is typically diagnosed based on the presenting signs and symptoms, in combination with supporting medical imaging and laboratory testing.[4] It can be difficult to confirm, especially early on, since the signs and symptoms may be similar to those of other medical problems.[5][6] The McDonald criteria, which focus on clinical, laboratory, and radiologic evidence of lesions at different times and in different areas, is the most commonly used method of diagnosis[7] with the Schumacher and Poser criteria being of mostly historical significance.[8] While the above criteria allow for a non-invasive diagnosis, some state that the only definitive proof is an autopsy or biopsy where lesions typical of MS are detected.[5][2][3]
Clinical data alone may be sufficient for a diagnosis of MS if an individual has had separate episodes of neurologic symptoms characteristic of the disease.[2] In those who seek medical attention after only one attack, other testing is needed for the diagnosis. The most commonly used diagnostic tools are neuroimaging, analysis of cerebrospinal fluid and evoked potentials. Magnetic resonance imaging of the brain and spine may show areas of demyelination (lesions or plaques). Gadolinium can be administered intravenously as a contrast agent to highlight active plaques and, by elimination, demonstrate the existence of historical lesions not associated with symptoms at the moment of the evaluation.[2][9] Testing of cerebrospinal fluid obtained from a lumbar puncture can provide evidence of chronic inflammation in the central nervous system. The cerebrospinal fluid is tested for oligoclonal bands of IgG on electrophoresis, which are inflammation markers found in 75–85% of people with MS.[2][10] The nervous system in MS may respond less actively to stimulation of the optic nerve and sensory nerves due to demyelination of such pathways. These brain responses can be examined using visual- and sensory-evoked potentials.[11]
Trends towards earlier diagnosis
Prodomal or early phase
Studies have found increased interactions with health services by people who were diagnosed with MS several years later. The range of symptoms included pain and fatigue. Studies continue on whether MS has a prodromal or early phase, and could possibly be diagnosed and treated much earlier.[12][13][14][15]
Diagnostic criteria
McDonald criteria
As of 2021, the McDonald criteria for MS are the most commonly used.
The 2017 McDonald criteria can be summarize in this table:
| Clinical Presentation | Additional Data Needed |
|---|---|
| * 2 or more attacks (relapses) * 2 or more objective clinical lesions |
None; clinical evidence will suffice (additional evidence desirable but must be consistent with MS) |
| * 2 or more attacks * 1 objective clinical lesion (as well as clear-cut historical evidence of a previous attack involving a lesion in a distinct anatomical location) |
None. |
| * 2 or more attacks * 1 objective clinical lesion |
Dissemination in space, demonstrated by an additional clinical attack implication a different CNS site or by MRI. |
| * 1 attack * 2 or more objective clinical lesions |
Dissemination in time, demonstrated by an additional clinical attack or by MRI
OR Demonstration of CSF-specific oligoclonal bands |
| * 1 attack * 1 objective clinical lesion (monosymptomatic presentation) |
Dissemination in space demonstrated by an additional clinical attack implicating a different CNS site or by MRI. AND Dissemination in time demonstrated by an additional clinical attack or by MRI, OR Demonstration of CSF-specific oligoclonal bands |
| Insidious neurological progression suggestive of MS (primary progressive MS) |
One year of disease progression (retrospectively or prospectively determined) and
Two of the following:
|
Okuda Criteria
These criteria, used mainly for research in MS, define what should be considered a Radiologically Isolated Syndrome (RIS).[18] Some reports point to the possibility of predicting RIS to CIS conversion based on oligoclonal bands and neurofilament light chain.[19]
Research into diagnostic techniques
Multiple sclerosis diagnosis can only be made when there is proof of lesions disseminated in time and in space. Therefore, when damage in the CNS is big enough to be seen. It would be desirable to make it faster.
The ideal diagnosis schema would be able to determine for any given subject, if he will develop MS, at any point in his life, and when. Nevertheless, not enough is currently known about the MS underlying conditions to achieve that.
In order to get as close as possible to the ideal diagnosis status, a lot of research into multiple sclerosis biomarkers is taking place currently.
Biomarkers in MS
An active field of research is looking for biomarkers for MS that could speed-up the diagnosis doing it more accurate at the same time. While most of them are still under research, there are some of them already well stablished:
- oligoclonal bands: They present proteins that are in the CNS or in blood. Those that are in CNS but not in blood suggest a diagnosis of MS.
- MRZ reaction: A polyspecific antiviral immune response against the viruses of measles, rubella and zoster found in 1992.[20] In some reports the MRZR showed a lower sensitivity than OCB (70% vs. 100%), but a higher specificity (69% vs. 92%) for MS.[20]
- free light chains (FLC), specially the kappa-FLCs (kFLCs). Several authors have reported that the nephelometric and ELISA FLCs determination is comparable with OCBs as markers of IgG synthesis, and kFLCs behave even better than oligoclonal bands.[21]
Differential diagnosis
Several conditions can mimic MS. Given the unknown pathogenesis of MS, its differential diagnosis is based in exclusion of known conditions.
Very close diseases with similar symptoms are the whole "inflammatory demyelinating diseases spectrum", specially neuromyelitis optica and anti-MOG associated encephalomyelitis.
Outside this spectrum, another important mimic is neuroborreliosis. A Borrelia-specific IgG index exists, and testing for it could make the differential diagnosis.[22]
Clinical courses
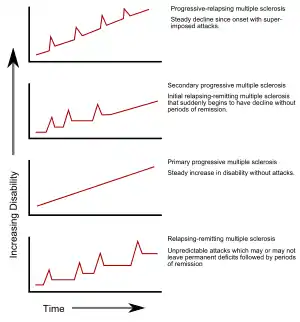
Several phenotypes (commonly named types), or patterns of progression, have been described. Phenotypes use the past course of the disease in an attempt to predict the future course. They are important not only for prognosis but also for treatment decisions. In 1996, the United States National Multiple Sclerosis Society described four clinical courses.[23]
The original structure approved in 1996, sometimes still used, was:
- relapsing-remitting
- secondary progressive (SPMS)
- primary progressive (PPMS)
- progressive relapsing.
This set of courses was reviewed by an international panel in 2013,[24][25] adding clinically isolated syndrome (CIS) and radiologically isolated syndrome (RIS) as phenotypes, and finally removing the "progressive relapsing" phenotype. They also added modificators for the clinical courses based on a pair of characteristics: Active/non-active and with/without progression.
The four currently accepted courses or stages are:
- Clinically isolated syndrome (CIS)
- Relapsing-remitting MS (RRMS)
- Primary progressive MS (PPMS)
- Secondary progressive MS (SPMS)
Relapsing-remitting
The relapsing-remitting subtype is characterized by unpredictable relapses followed by periods of months to years of relative quiet (remission) with no new signs of disease activity. Deficits that occur during attacks may either resolve or leave problems, the latter in about 40% of attacks and being more common the longer a person has had the disease.[4][5] This describes the initial course of 80% of individuals with MS.[5] When deficits always resolve between attacks, this is sometimes referred to as benign MS,[26] although people will still build up some degree of disability in the long term.[5] On the other hand, the term malignant multiple sclerosis is used to describe people with MS having reached significant level of disability in a short period.[27] The relapsing-remitting subtype usually begins with a clinically isolated syndrome (CIS). In CIS, a person has an attack suggestive of demyelination, but does not fulfill the criteria for multiple sclerosis.[5][28] 30 to 70% of persons experiencing CIS later develop MS.[28]
Secondary progressive
Secondary progressive MS occurs in around 65% of those with initial relapsing-remitting MS, who eventually have progressive neurologic decline between acute attacks without any definite periods of remission.[5][23] Occasional relapses and minor remissions may appear.[23] The most common length of time between disease onset and conversion from relapsing-remitting to secondary progressive MS is 19 years.[29]
Primary progressive
The primary progressive subtype occurs in approximately 10–20% of individuals, with no remission after the initial symptoms.[4][30] It is characterized by progression of disability from onset, with no, or only occasional and minor, remissions and improvements.[23] The usual age of onset for the primary progressive subtype is later than of the relapsing-remitting subtype. It is similar to the age that secondary progressive usually begins in relapsing-remitting MS, around 40 years of age.[5]
Progressive relapsing
Progressive relapsing MS describes those individuals who, from onset, have a steady neurologic decline but also have clear superimposed attacks. This is the least common of all subtypes.[23]
Atypical MS
Unusual types of MS have been described; these include Devic's disease, Balo concentric sclerosis, Schilder's diffuse sclerosis, and Marburg multiple sclerosis. There is debate on whether they are MS variants or different diseases.[31] Multiple sclerosis behaves differently in children, taking more time to reach the progressive stage.[5] Nevertheless, they still reach it at a lower average age than adults usually do.[5]
History
Since the first description of multiple sclerosis (MS) by Charcot, the Neurological community has been striving to create reliable and reproducible criteria for diagnosis of MS.[32] The first attempts were made by Charcot himself, followed by Marburg and later Allison. The first criteria however were lacking in sensitivity and specificity for clinical use.[32]
The first landmark event in the history of diagnostic criteria for MS was the development of the Schumacher criteria. These were the first internationally recognized criteria for diagnosis of MS and introduced very important diagnostic concepts that are the cornerstone of MS diagnosis nowadays, such as the clinical definition of MS and the requirement of dissemination in time and space for accurate diagnosis.
Since then, other diagnostic criteria have been proposed. Among them, Poser criteria utilized several laboratory and paraclinical studies to enhance the diagnostic accuracy. McDonald criteria, which are the ones used today, successfully introduced MRI findings as surrogates for the criterion of dissemination in time and space when clinical data are lacking, thus allowing earlier diagnosis of MS.[32]
Schumacher criteria
To get a diagnosis of CDMS a patient must show the following:[33]
- Clinical signs of a problem in the CNS
- Evidence of two or more areas of CNS involvement
- Evidence of white matter involvement
- One of these: Two or more relapses (each lasting ≥ 24 hr and separated by at least 1 month) or progression (slow or stepwise)
- Patient should be between 10 and 50 yr old at time of examination
- No better explanation for patient's symptoms and signs
The last condition, no better explanation for symptoms, has been heavily criticised, but it has been preserved and it is currently included in the new McDonalds criteria in the form that "no better explanation should exist for MRI observations"
Poser criteria
Poser criteria can be summarized in this table:
Any of the five conclusions have subpossibilities. Here a table is shown with each one of them:
| Clinical Presentation | Additional Data Needed | |
|---|---|---|
| CDMS | * Two or more attacks (relapses) | Two clinical evidence One clinical and one paraclinical evidence |
| LSDMS | * At least one attack and oligoclonal bands | Two attacks and one evidence (clinical or paraclinical) One attack and two clinical evidences One attack, one clinical and one paraclinical evidences |
| CPMS | * At least one attack | Two attacks and one clinical evidence One attack and two clinical evidences One attack, one clinical and one paraclinical evidences |
| LSPMS | * Two attacks | No more evidence is required |
If none of these requirements is fulfilled, the diagnosis is "No MS", meaning that there is not enough clinical evidence to support a clinical diagnosis of MS.
Barkhof-Tintoré criteria
Barkhof criteria,[34] later modified by Tintoré[35] were an early attempt to use MRI to diagnose MS. It was developed by Frederik Barkhof.[34]
Their observations were taken into account when McDonald criteria were published, and therefore they can be considered deprecated by the latter.
References
- Oki S (February 2018). "Novel mechanism and biomarker of chronic progressive multiple sclerosis". Clinical and Experimental Neuroimmunology. 9 (1): 25–34. doi:10.1111/cen3.12449.
- McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, et al. (July 2001). "Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis". Annals of Neurology. 50 (1): 121–127. CiteSeerX 10.1.1.466.5368. doi:10.1002/ana.1032. PMID 11456302. S2CID 13870943.
- Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L, et al. (December 2005). "Diagnostic criteria for multiple sclerosis: 2005 revisions to the "McDonald Criteria"". Annals of Neurology. 58 (6): 840–846. CiteSeerX 10.1.1.604.2677. doi:10.1002/ana.20703. PMID 16283615. S2CID 54512368.
- Tsang BK, Macdonell R (December 2011). "Multiple sclerosis- diagnosis, management and prognosis". Australian Family Physician. 40 (12): 948–955. PMID 22146321.
- Compston A, Coles A (October 2008). "Multiple sclerosis". Lancet. 372 (9648): 1502–1517. doi:10.1016/S0140-6736(08)61620-7. PMID 18970977. S2CID 195686659.
- Trojano M, Paolicelli D (November 2001). "The differential diagnosis of multiple sclerosis: classification and clinical features of relapsing and progressive neurological syndromes". Neurological Sciences. 22 (8 Suppl 2): S98-102. doi:10.1007/s100720100044. PMID 11794488. S2CID 3057096.
- Atlas: Multiple Sclerosis Resources in the World, 2008. World Health Organization & Multiple Sclerosis International Federation. 2008. pp. 15–16. hdl:10665/43968. ISBN 978-92-4-156375-8.
- Poser CM, Brinar VV (June 2004). "Diagnostic criteria for multiple sclerosis: an historical review". Clinical Neurology and Neurosurgery. 106 (3): 147–158. doi:10.1016/j.clineuro.2004.02.004. PMID 15177763. S2CID 23452341.
- Rashid W, Miller DH (February 2008). "Recent advances in neuroimaging of multiple sclerosis". Seminars in Neurology. 28 (1): 46–55. doi:10.1055/s-2007-1019127. PMID 18256986.
- Link H, Huang YM (November 2006). "Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness". Journal of Neuroimmunology. 180 (1–2): 17–28. doi:10.1016/j.jneuroim.2006.07.006. PMID 16945427. S2CID 22724352.
- Gronseth GS, Ashman EJ (May 2000). "Practice parameter: the usefulness of evoked potentials in identifying clinically silent lesions in patients with suspected multiple sclerosis (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology". Neurology. 54 (9): 1720–1725. doi:10.1212/wnl.54.9.1720. PMID 10802774.
- Tremlett H, Okuda DT, Lebrun-Frenay C (October 2021). "The multiple sclerosis prodrome is just unspecific symptoms in radiologically isolated syndrome patients - No". Multiple Sclerosis. 27 (12): 1824–1826. doi:10.1177/13524585211035951. PMC 8521363. PMID 34494923.
- Yusuf F, Wijnands JM, Kingwell E, Zhu F, Evans C, Fisk JD, et al. (February 2021). "Fatigue, sleep disorders, anaemia and pain in the multiple sclerosis prodrome". Multiple Sclerosis. 27 (2): 290–302. doi:10.1177/1352458520908163. PMID 32250183. S2CID 214810456.
- Disanto G, Zecca C, MacLachlan S, Sacco R, Handunnetthi L, Meier UC, et al. (June 2018). "Prodromal symptoms of multiple sclerosis in primary care". Annals of Neurology. 83 (6): 1162–1173. doi:10.1002/ana.25247. PMID 29740872. S2CID 13691058.
- Tremlett H, Marrie RA (January 2021). "The multiple sclerosis prodrome: Emerging evidence, challenges, and opportunities". Multiple Sclerosis. 27 (1): 6–12. doi:10.1177/1352458520914844. PMID 32228281. S2CID 214750591.
- Spelman T, Magyari M, Piehl F, Svenningsson A, Rasmussen PV, Kant M, et al. (October 2021). "Treatment Escalation vs Immediate Initiation of Highly Effective Treatment for Patients With Relapsing-Remitting Multiple Sclerosis: Data From 2 Different National Strategies". JAMA Neurology. 78 (10): 1197–1204. doi:10.1001/jamaneurol.2021.2738. PMC 8369379. PMID 34398221. S2CID 237095594.
- Claflin SB, Campbell JA, Mason DF, Kalincik T, Simpson-Yap S, Norman R, et al. (August 2021). "The effect of national disease-modifying therapy subsidy policy on long-term disability outcomes in people with multiple sclerosis". Multiple Sclerosis. 28 (5): 831–841. doi:10.1177/13524585211035948. PMID 34387513. S2CID 236997752.
- Okuda DT, Mowry EM, Beheshtian A, Waubant E, Baranzini SE, Goodin DS, et al. (March 2009). "Incidental MRI anomalies suggestive of multiple sclerosis: the radiologically isolated syndrome". Neurology. 72 (9): 800–805. doi:10.1212/01.wnl.0000335764.14513.1a. PMID 19073949. S2CID 9981947.
- Matute-Blanch C, Villar LM, Álvarez-Cermeño JC, Rejdak K, Evdoshenko E, Makshakov G, et al. (April 2018). "Neurofilament light chain and oligoclonal bands are prognostic biomarkers in radiologically isolated syndrome". Brain. 141 (4): 1085–1093. doi:10.1093/brain/awy021. PMID 29452342.
- Hottenrott T, Dersch R, Berger B, Rauer S, Eckenweiler M, Huzly D, Stich O (December 2015). "The intrathecal, polyspecific antiviral immune response in neurosarcoidosis, acute disseminated encephalomyelitis and autoimmune encephalitis compared to multiple sclerosis in a tertiary hospital cohort". Fluids and Barriers of the CNS. 12 (1): 27. doi:10.1186/s12987-015-0024-8. PMC 4677451. PMID 26652013.
- Fabio Duranti; Massimo Pieri; Rossella Zenobi; Diego Centonze; Fabio Buttari; Sergio Bernardini; Mariarita Dessi (August 2015). "kFLC Index: a novel approach in early diagnosis of Multiple Sclerosis". International Journal of Scientific Research. 4 (8).
- Kotulska-Jóźwiak K, Pacheva I, Patrova A, Jurkiewicz EJ, Ivanov I, Kuczyński D, Geneva I (18 February 2019). "Lyme Disease or Multiple Sclerosis? Two cases with overlapping features". Journal of the International Child Neurology Association: 12. doi:10.17724/jicna.2018.112. S2CID 149606491.
- Lublin FD, Reingold SC (April 1996). "Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis". Neurology. 46 (4): 907–911. doi:10.1212/WNL.46.4.907. PMID 8780061. S2CID 40213123.
- Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, et al. (July 2014). "Defining the clinical course of multiple sclerosis: the 2013 revisions". Neurology. 83 (3): 278–286. doi:10.1212/WNL.0000000000000560. PMC 4117366. PMID 24871874.
- National Multiple Sclerosis Society. "Changes in multiple sclerosis disease-course (or "type") descriptions" (PDF). Archived (PDF) from the original on 3 August 2016. Retrieved 21 August 2017.
NEW COURSE ADDED: Clinically Isolated Syndrome (CIS)...COURSE ELIMINATED: Progressive Relapsing (PRMS).
- Pittock SJ, Rodriguez M (2008). "Benign Multiple Sclerosis: A Distinct Clinical Entity with Therapeutic Implications". Advances in multiple Sclerosis and Experimental Demyelinating Diseases. Current Topics in Microbiology and Immunology. Vol. 318. pp. 1–17. doi:10.1007/978-3-540-73677-6_1. ISBN 978-3-540-73676-9. PMID 18219812.
- Feinstein A (2007). The Clinical Neuropsychiatry of Multiple Sclerosis. Cambridge University Press. p. 20. ISBN 978-0-521-85234-0.
- Miller D, Barkhof F, Montalban X, Thompson A, Filippi M (May 2005). "Clinically isolated syndromes suggestive of multiple sclerosis, part I: natural history, pathogenesis, diagnosis, and prognosis". The Lancet. Neurology. 4 (5): 281–288. doi:10.1016/S1474-4422(05)70071-5. PMID 15847841. S2CID 36401666.
- Rovaris M, Confavreux C, Furlan R, Kappos L, Comi G, Filippi M (April 2006). "Secondary progressive multiple sclerosis: current knowledge and future challenges". The Lancet. Neurology. 5 (4): 343–354. doi:10.1016/S1474-4422(06)70410-0. PMID 16545751. S2CID 39503553.
- Miller DH, Leary SM (October 2007). "Primary-progressive multiple sclerosis". The Lancet. Neurology. 6 (10): 903–912. doi:10.1016/S1474-4422(07)70243-0. hdl:1871/24666. PMID 17884680. S2CID 31389841.
- Stadelmann C, Brück W (November 2004). "Lessons from the neuropathology of atypical forms of multiple sclerosis". Neurological Sciences. 25 (Suppl 4): S319–S322. doi:10.1007/s10072-004-0333-1. PMID 15727225. S2CID 21212935.
- Ntranos A, Lublin F (October 2016). "Diagnostic Criteria, Classification and Treatment Goals in Multiple Sclerosis: The Chronicles of Time and Space". Current Neurology and Neuroscience Reports. 16 (10): 90. doi:10.1007/s11910-016-0688-8. PMID 27549391. S2CID 4498332.
- Marriott JJ, O'Connor PW (2010). "Differential Diagnosis and Diagnostic Criteria for Multiple Sclerosis". Multiple Sclerosis 3. Blue Books of Neurology. Vol. 35. pp. 19–42. doi:10.1016/B978-1-4160-6068-0.00002-4. ISBN 978-1-4160-6068-0. S2CID 68141248.
- Barkhof F, Filippi M, Miller DH, Scheltens P, Campi A, Polman CH, et al. (November 1997). "Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis". Brain. 120 (11): 2059–2069. doi:10.1093/brain/120.11.2059. PMID 9397021.
- Tintoré M, Rovira A, Martínez MJ, Rio J, Díaz-Villoslada P, Brieva L, et al. (April 2000). "Isolated demyelinating syndromes: comparison of different MR imaging criteria to predict conversion to clinically definite multiple sclerosis". AJNR. American Journal of Neuroradiology. 21 (4): 702–706. PMC 7976636. PMID 10782781.