Treacher Collins syndrome
Treacher Collins syndrome (TCS) is a genetic disorder characterized by deformities of the ears, eyes, cheekbones, and chin.[5] The degree to which a person is affected, however, may vary from mild to severe.[5] Complications may include breathing problems, problems seeing, cleft palate, and hearing loss.[5] Those affected generally have normal intelligence.[5]
| Treacher Collins syndrome | |
|---|---|
| Other names | Treacher Collins–Franceschetti syndrome,[1] mandibulofacial dysostosis,[2] Franceschetti-Zwalen-Klein syndrome[3] |
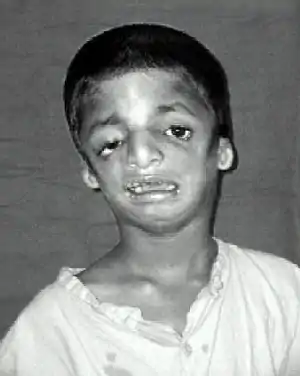 | |
| Child with Treacher Collins syndrome[4] | |
| Specialty | Medical genetics |
| Symptoms | Deformities of the ears, eyes, cheekbones, chin[5] |
| Complications | Breathing problems, problems seeing, hearing loss[5] |
| Causes | Genetic[5] |
| Diagnostic method | Based on symptoms, X-rays, genetic testing[3] |
| Differential diagnosis | Nager syndrome, Miller syndrome, hemifacial microsomia[3] |
| Treatment | Reconstructive surgery, hearing aids, speech therapy[6] |
| Prognosis | Generally normal life expectancy[6] |
| Frequency | 1 in 50,000 people[5] |
TCS is usually autosomal dominant.[5] More than half the time it occurs as a result of a new mutation rather than being inherited from a person's parents.[5] The involved genes may include TCOF1, POLR1C, or POLR1D.[5] Diagnosis is generally suspected based on symptoms and X-rays, and potentially confirmation by genetic testing.[3]
Treacher Collins syndrome is not curable.[6] Symptoms may be managed with reconstructive surgery, hearing aids, speech therapy, and other assistive devices.[6] Life expectancy is generally normal.[6] TCS occurs in about one in 50,000 people.[5] The syndrome is named after Edward Treacher Collins, an English surgeon and ophthalmologist, who described its essential traits in 1900.[7][8]
Signs and symptoms
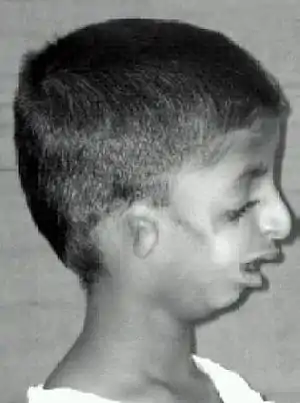
Symptoms in people with Treacher Collins syndrome vary. Some individuals are so mildly affected that they remain undiagnosed, while others have moderate to severe facial involvement and life-threatening airway compromise.[9] Most of the features of TCS are symmetrical and are already recognizable at birth.
The most common symptom of Treacher Collins syndrome is underdevelopment of the lower jaw and underdevelopment of the zygomatic bone. This can be accompanied by the tongue being retracted. The small mandible can result in a poor occlusion of the teeth or in more severe cases, trouble breathing or swallowing. The respiratory system of a child with Treacher Collins syndrome is the primary concern at birth, with other issues only addressed once respiratory function has been stabilized.[10] Underdevelopment of the zygomatic bone gives the cheeks a sunken appearance.[11][12]
The external ear is sometimes small, rotated, malformed, or absent entirely in people with TCS. Symmetric, bilateral narrowing or absence of the external ear canal, is also described.[12][13] In most cases, the bones of the middle ear and the middle ear cavity are misshapen. Inner ear malformations are rarely described. As a result of these abnormalities, a majority of the individuals with TCS have conductive hearing loss.[12][14]
Most affected people also experience eye problems, including coloboma (notches) in the lower eyelids, partial or complete absence of eyelashes on the lower lid, downward angled eyelids, drooping of upper and lower eyelids, and narrowing of the tear ducts. Vision loss can occur and is associated with strabismus, refractive errors, and anisometropia. It can also be caused by severely dry eyes, a consequence of lower eyelid abnormalities and frequent eye infections.[12][13][15][16]
Although an abnormally shaped skull is not distinctive for Treacher Collins syndrome, brachycephaly with bitemporal narrowing is sometimes observed.[13] Cleft palate is also common.[12]
Dental anomalies are seen in 60% of affected people, including tooth agenesis (33%), discoloration (enamel opacities) (20%), malplacement of the maxillary first molars (13%), and wide spacing of the teeth. In some cases, dental anomalies in combination with mandible hypoplasia result in a malocclusion. This can lead to problems with food intake and the ability to close the mouth.[12]
Less common features of TCS may add to an affected person's breathing problems, including sleep apnea. Choanal atresia or stenosis is a narrowing or absence of the choanae, the internal opening of the nasal passages, which may also be observed. Underdevelopment of the pharynx can also narrow the airway.[12]
Features related to TCS that are seen less frequently include nasal deformities, high-arched palate, macrostomia, preauricular hair displacement, cleft palate, hypertelorism, notched upper eyelid, and congenital heart defects.[11][12][16]
Although facial deformity is often associated with developmental delay and intellectual disability, more than 95% of people affected with TCS have normal intelligence.[12] The psychological and social problems associated with facial deformity can affect quality of life in individuals with TCS.
Genetics
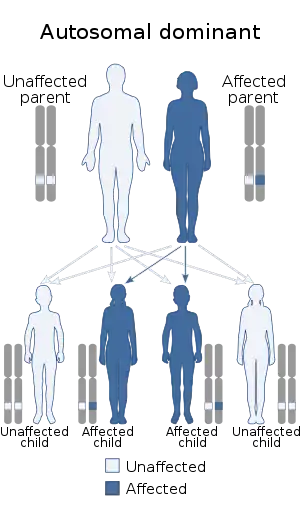
Mutations in TCOF1, POLR1C, or POLR1D genes can cause Treacher Collins syndrome.[17] TCOF1 gene mutations are the most common cause of the disorder, with POLR1C and POLR1D gene mutations causing an additional 2% of cases. In individuals without an identified mutation in one of these genes, the genetic cause of the condition is unknown. The TCOF1, POLR1C, and POLR1D genes code for proteins which play important roles in the early development of bones and other tissues of the face. Mutations in these genes reduce the production of rRNA, which may trigger the self-destruction (apoptosis) of certain cells involved in the development of facial bones and tissues. It is unclear why the effects of a reduction in rRNA are limited to facial development. Mutations in TCOF1 and POLR1D cause the autosomal dominant form of Treacher Collins, and mutations in POLR1C cause the autosomal recessive form.[12]
TCOF1
TCOF1 is the primary gene associated with TCS, a mutation in this gene being found in 90–95% of the individuals with TCS.[11][18] However, in some individuals with typical symptoms of TCS, mutations in TCOF1 have not been found.[19] Investigation of the DNA has resulted in the identification of the kind of mutations found in TCOF1. The majority of mutations are small deletions or insertions, though splice site and missense mutations also have been identified.[11][20][21][22]
Mutation analysis has unveiled more than 100 disease-causing mutations in TCOF1, which are mostly family-specific mutations. The only recurrent mutation accounts for about 17% of the cases.[23]
TCOF1 is found on the 5th chromosome in the 5q32 region. It codes for a relatively simple nucleolar protein called treacle, that is thought to be involved in ribosome assembly.[18] Mutations in TCOF1 lead to haploinsufficiency of the treacle protein.[24] Haploinsufficiency occurs when a diploid organism has only one functional copy of a gene, because the other copy is inactivated by a mutation. The one normal copy of the gene does not produce enough protein, causing disease. Haploinsufficiency of the treacle protein leads to a depletion of the neural crest cell precursor, which leads to a reduced number of crest cells migrating to the first and second pharyngeal arches. These cells play an important role in the development of the craniofacial appearance, and loss of one copy of treacle affects the cells' ability to form the bones and tissues of the face.[12][20][25]
Other mutations
POLR1C and POLR1D mutations are responsible for a minority of cases of Treacher Collins. POLR1C is found on chromosome 6 at position 6q21.2 and POLR1D is found on chromosome 13 at position 13q12.2. Those genes code for a protein subunits shared between RNA polymerase I and III. Both of these polymerases are important for ribosome biogenesis.[12]
Diagnosis
Genetic counseling
TCS is inherited in an autosomal dominant manner and the penetrance of the affected gene is almost complete.[26] Some recent investigations, though, described some rare cases in which the penetrance in TCS was not complete. Causes may be a variable expressivity, an incomplete penetrance[27] or germline mosaicism.[28] Only 40% of the mutations are inherited. The remaining 60% are a result of a de novo mutation, where a child has a new mutation in the responsible gene and did not inherit it from either parent.[12][29] In the outcome of the disease, inter- and intrafamilial variability occurs. This suggests that when an affected child is born, it is important to investigate the parents to determine whether the affected gene is present, because the parent could have a mild form of the disease that has not been diagnosed. In this case, the risk of having another affected child is 50%. If the parents do not have the affected gene, the recurrence risk appears to be low.[26] In following generations, the severity of the clinical symptoms increases.[21]
Prenatal diagnosis
Mutations in the main genes responsible for TCS can be detected with chorionic villus sampling or amniocentesis. Rare mutations may not be detected by these methods. Ultrasonography can be used to detect craniofacial abnormalities later in pregnancy, but may not detect milder cases.[12]
Clinical findings
TCS is often first suspected with characteristic symptoms observed during a physical exam. However, the clinical presentation of TCS can resemble other diseases, making diagnosis difficult.[30] The OMENS classification was developed as a comprehensive and stage-based approach to differentiate the diseases. This acronym describes five distinct dysmorphic manifestations, namely orbital asymmetry, mandibular hypoplasia, auricular deformity, nerve development, and soft-tissue disease.[31]
Orbital symmetry
- O0: normal orbital size, position
- O1: abnormal orbital size
- O2: abnormal orbital position
- O3: abnormal orbital size and position
Mandible
- M0: normal mandible
- M1: small mandible and glenoid fossa with short ramus
- M2: ramus short and abnormally shaped
- 2A: glenoid fossa in anatomical acceptable position
- 2B: Temperomandibular joint inferiorly (TMJ), medially, anteriorly displaced, with severely hypoplastic condyle
- M3: Complete absence of ramus, glenoid fossa, and TMJ
Ear
- E0: normal ear
- E1: Minor hypoplasia and cupping with all structures present
- E2: Absence of external auditory canal with variable hypoplasia of the auricle
- E3: Malposition of the lobule with absent auricle, lobular remnant usually inferior anteriorly displaced
Facial nerve
- N0: No facial nerve involvement
- N1: Upper facial nerve involvement (temporal or zygomatic branches)
- N2: Lower facial nerve involvement (buccal, mandibular or cervical)
- N3: All branches affected
Soft tissue
- S0: No soft tissue or muscle deficiency
- S1: Minimal tissue or muscle deficiency
- S2: Moderate tissue or muscle deficiency
- S3: Severe tissue or muscle deficiency
Radiographs
A few techniques are used to confirm the diagnosis in TCS.[30][32]
An orthopantomogram (OPG) is a panoramic dental X-ray of the upper and lower jaw. It shows a two-dimensional image from ear to ear. Particularly, OPG facilitates an accurate postoperative follow-up and monitoring of bone growth under a mono- or double-distractor treatment. Thereby, some TCS features could be seen on OPG, but better techniques are used to include the whole spectrum of TCS abnormalities instead of showing only the jaw abnormalities.[30]
Another method of radiographic evaluation is taking an X-ray image of the whole head. The lateral cephalometric radiograph in TCS shows hypoplasia of the facial bones, like the malar bone, mandible, and the mastoid.[30]
Finally, occipitomental radiographs are used to detect hypoplasia or discontinuity of the zygomatic arch.[32]
CT scan
A temporal-bone CT using thin slices makes it possible to diagnose the degree of stenosis and atresia of the external auditory canal, the status of the middle ear cavity, the absent or dysplastic and rudimentary ossicles, or inner ear abnormalities such as a deficient cochlea. Two- and three-dimensional CT reconstructions with VRT and bone and skin-surfacing are helpful for more accurate staging and the three-dimensional planning of mandibular and external ear reconstructive surgery.
Differential diagnosis
Other diseases have similar characteristics to Treacher Collins syndrome. In the differential diagnosis, one should consider the acrofacial dysostoses. The facial appearance resembles that of Treacher Collins syndrome, but additional limb abnormalities occur in those persons. Examples of these diseases are Nager syndrome and Miller syndrome.
The oculoauriculovertebral spectrum should also be considered in the differential diagnosis. An example is hemifacial microsomia, which primarily affects development of the ear, mouth, and mandible. This anomaly may occur bilaterally. Another disease which belongs to this spectrum is Goldenhar syndrome, which includes vertebral abnormalities, epibulbar dermoids and facial deformities.[33]
Treatment
The treatment of individuals with TCS may involve the intervention of professionals from multiple disciplines. The primary concerns are breathing and feeding, as a consequence of the hypoplasia of the mandibula and the obstruction of the hypopharynx by the tongue. Sometimes, they may require a tracheostomy to maintain an adequate airway,[34] and a gastrostomy to assure an adequate caloric intake while protecting the airway. Corrective surgery of the face is performed at defined ages, depending on the developmental state.[35]
An overview of the present guidelines:
- If a cleft palate is present, the repair normally takes place at 9–12 months old. Before surgery, a polysomnography with a palatal plate in place is needed. This may predict the postoperative situation and gives insight on the chance of the presence of sleep apnea (OSAS) after the operation.[11][36][37]
- Hearing loss is treated by bone conduction amplification, speech therapy, and educational intervention to avoid language/speech problems. The bone-anchored hearing aid is an alternative for individuals with ear anomalies.[38]
- Zygomatic and orbital reconstruction is performed when the cranio-orbitozygomatic bone is completely developed, usually at the age of 5–7 years. In children, an autologous bone graft is mostly used. In combination with this transplantation, lipofilling can be used in the periorbital area to get an optimal result of the reconstruction. Reconstruction of the lower eyelid coloboma includes the use of a myocutaneous flap, which is elevated and in this manner closes the eyelid defect.[39]
- External ear reconstruction is usually done when the individual is at least eight years old. Sometimes, the external auditory canal or middle ear can also be treated.
- The optimal age for the maxillomandibular reconstruction is controversial; as of 2004, this classification has been used:[11]
- Type I (mild) and Type IIa (moderate) 13–16 years
- Type IIb (moderate to severe malformation) at skeletal maturity
- Type III (severe) 6–10 years
- When the teeth are cutting, the teeth should be under supervision of an orthodontist to make sure no abnormalities occur. If abnormalities like dislocation or an overgrowth of teeth are seen, appropriate action can be undertaken as soon as possible.[20]
- Orthognathic treatments usually take place after the age of 16 years; at this point, all teeth are in place and the jaw and dentition are mature. Whenever OSAS is detected, the level of obstruction is determined through endoscopy of the upper airways. Mandibular advancement can be an effective way to improve both breathing and æsthetics, while a genioplasty only restores the profile.[11]
- If a nose reconstruction is necessary, it is usually performed after the orthognathic surgery and after the age of 18 years.[11]
- The contour of the facial soft tissues generally requires correction at a later age, because of the facial skeletal maturity. The use of microsurgical methods, like the free flap transfer, has improved the correction of facial soft tissue contours.[40] Another technique to improve the facial soft tissue contours is lipofilling. For instance, lipofilling is used to reconstruct the eyelids.[39]
Hearing loss
Hearing loss in Treacher Collins syndrome is caused by deformed structures in the outer and middle ear. The hearing loss is generally bilateral with a conductive loss of about 50–70 dB. Even in cases with normal auricles and open external auditory canals, the ossicular chain is often malformed.[41]
Attempts to surgically reconstruct the external auditory canal and improve hearing in children with TCS have not yielded positive results.[42]
Auditory rehabilitation with bone-anchored hearing aids (BAHAs) or a conventional bone conduction aid has proven preferable to surgical reconstruction.[38]
Psychiatric
The disorder can be associated with a number of psychological symptoms, including anxiety, depression, social phobia, and distress about body image. People who have this disorder may also experience discrimination, bullying, and name calling, especially when young. A multi-disciplinary team and parental support should include these issues.[43]
Epidemiology
TCS occurs in about one in 50,000 births in Europe.[44] Worldwide, it is estimated to occur in one in 10,000 to one in 50,000 births.[12]
History
The syndrome is named after Edward Treacher Collins (1862–1932), the English surgeon and ophthalmologist who described its essential traits in 1900.[7][8][45] In 1949, Adolphe Franceschetti and David Klein described the same condition on their own observations as mandibulofacial dysostosis. The term mandibulofacial dysostosis is used to describe the clinical features.[46]
Culture
A July 1977 New York Times article[47] that was reprinted in numerous newspapers nationwide over the ensuing weeks brought this malady to many people's attention for the first time.
Prior to beginning his comedy career, Bob Saget made a documentary short called "Through Adam's Eyes" documenting his young nephew's experiences undergoing facial reconstructive surgery due to Treacher Collins; the film won a student Academy Award. [48]
The disorder was featured on the show Nip/Tuck, in the episode "Blu Mondae".[49] TLC's Born Without a Face[50] features Juliana Wetmore, who was born with the most severe case in medical history of this syndrome and is missing 30%–40% of the bones in her face.[50]
In 2010, BBC Three documentary Love Me, Love My Face[51] covered the case of a man, Jono Lancaster, with the condition. In 2011, BBC Three returned to Jono to cover his and his partner Laura's quest to start a family,[2] in So What If My Baby Is Born Like Me?,[52] which first aired as part of a BBC Three season of programmes on parenting.[53] The first film was replayed on BBC One shortly ahead of the second film's initial BBC Three broadcast. Lancaster's third BBC Three film, Finding My Family on Facebook, which looked at adoption, aired in 2011.[54]
In Wonder, a children's novel, the main character is a child who has Treacher Collins syndrome.[55] A 2017 film adaptation of Wonder, starring Julia Roberts, Owen Wilson and Jacob Tremblay, was released in November 2017.[56][57]
Alison Midstokke, who appears in the drama film Happy Face (2018),[58] is an actress and activist who has the condition.
References
- Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. pp. 894, 1686. ISBN 978-1-4160-2999-1.
- "I hated seeing my face in the mirror". BBC Online. 18 November 2010. Archived from the original on 2018-10-11. Retrieved 2010-11-18.
- "Treacher Collins Syndrome". NORD (National Organization for Rare Disorders). 2016. Archived from the original on 20 December 2019. Retrieved 7 November 2017.
- Goel, L; Bennur, SK; Jambhale, S (August 2009). "Treacher-collins syndrome-a challenge for anaesthesiologists". Indian Journal of Anaesthesia. 53 (4): 496–500. PMC 2894488. PMID 20640217.
- "Treacher Collins syndrome". Genetics Home Reference. June 2012. Archived from the original on 26 June 2020. Retrieved 7 November 2017.
- "Treacher Collins syndrome". rarediseases.info.nih.gov. 2015. Archived from the original on 16 August 2019. Retrieved 7 November 2017.
- R, Pramod John; John, Pramod (2014). Textbook of Oral Medicine. JP Medical Ltd. p. 76. ISBN 9789350908501. Archived from the original on 2020-09-12. Retrieved 2017-09-17.
- Beighton, Greta (2012). The Man Behind the Syndrome. Springer Science & Business Media. p. 173. ISBN 9781447114154.
- Edwards, S J; Fowlie, A; Cust, M P; Liu, D T; Young, I D; Dixon, M J (1 July 1996). "Prenatal diagnosis in Treacher Collins syndrome using combined linkage analysis and ultrasound imaging". Journal of Medical Genetics. 33 (7): 603–606. doi:10.1136/jmg.33.7.603. PMC 1050672. PMID 8818950.
- Mouthon, L., Busa, T., Bretelle, F., Karmous‐Benailly, H., Missirian, C., Philip, N., & Sigaudy, S. (2019). Prenatal diagnosis of micrognathia in 41 fetuses: Retrospective analysis of outcome and genetic etiologies. American Journal of Medical Genetics Part A, 179(12), 2365–2373. doi: 10.1002/ajmg.a.61359
- Katsanis SH, et al., Treacher Collins syndrome, 2004, GeneReviews
- "The Physician's Guide to Treacher Collins Syndrome" (PDF). National Organization for Rare Disorders (NORD). 2012. Archived from the original (PDF) on 2017-01-28.
- Posnick, Jeffrey C (1 October 1997). "Treacher Collins syndrome: Perspectives in evaluation and treatment". Journal of Oral and Maxillofacial Surgery. 55 (10): 1120–1133. doi:10.1016/S0278-2391(97)90294-9. PMID 9331237.
- Trainor, Paul A; Dixon, Jill; Dixon, Michael J (24 December 2008). "Treacher Collins syndrome: etiology, pathogenesis and prevention". European Journal of Human Genetics. 17 (3): 275–283. doi:10.1038/ejhg.2008.221. PMC 2986179. PMID 19107148.
- Hertle, R W; Ziylan, S; Katowitz, J A (1 October 1993). "Ophthalmic features and visual prognosis in the Treacher-Collins syndrome". British Journal of Ophthalmology. 77 (10): 642–645. doi:10.1136/bjo.77.10.642. PMC 504607. PMID 8218033.
- Marszałek, B; Wójcicki, P; Kobus, K; Trzeciak, WH (2002). "Clinical features, treatment and genetic background of Treacher Collins syndrome". Journal of Applied Genetics. 43 (2): 223–33. PMID 12080178.
- "Treacher Collins Syndrome". NORD (National Organization for Rare Disorders). Archived from the original on 2019-12-20. Retrieved 2016-02-29.
- Dixon, Jill; Edwards, Sara J.; Gladwin, Amanda J.; Dixon, Michael J.; Loftus, Stacie K.; Bonner, Cynthia A.; Koprivnikar, Kathryn; Wasmuth, John J. (31 January 1996). "Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome". Nature Genetics. 12 (2): 130–136. doi:10.1038/ng0296-130. PMID 8563749. S2CID 34312227.
- Teber OA, Gillessen-Kaesbach G, Fischer S, Böhringer S, Albrecht B, Albert A, Arslan-Kirchner M, Haan E, Hagedorn-Greiwe M, Hammans C, Henn W, Hinkel GK, König R, Kunstmann E, Kunze J, Neumann LM, Prott EC, Rauch A, Rott HD, Seidel H, Spranger S, Sprengel M, Zoll B, Lohmann DR, Wieczorek D (2004). "Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation". European Journal of Human Genetics. 12 (11): 879–90. doi:10.1038/sj.ejhg.5201260. PMID 15340364.
- Dixon, J; Trainor, P.; Dixon, M. J. (1 May 2007). "Treacher Collins syndrome". Orthodontics & Craniofacial Research. 10 (2): 88–95. doi:10.1111/j.1601-6343.2007.00388.x. PMID 17552945.
- Masotti, Cibele; Ornelas, Camila C.; Splendore-Gordonos, Alessandra; Moura, Ricardo; Félix, Têmis M.; Alonso, Nivaldo; Camargo, Anamaria A.; Passos-Bueno, Maria (1 January 2009). "Reduced transcription of TCOF1 in adult cells of Treacher Collins syndrome patients". BMC Medical Genetics. 10 (1): 136. doi:10.1186/1471-2350-10-136. PMC 2801500. PMID 20003452.
- Sakai, Daisuke; Trainor, Paul A. (31 May 2009). "Treacher Collins syndrome: Unmasking the role of Tcof1/treacle". The International Journal of Biochemistry & Cell Biology. 41 (6): 1229–1232. doi:10.1016/j.biocel.2008.10.026. PMC 3093759. PMID 19027870.
- Splendore, Alessandra; Fanganiello, Roberto D.; Masotti, Cibele; Morganti, Lucas S. C.; Rita Passos-Bueno, M. (1 May 2005). "TCOF1 mutation database: Novel mutation in the alternatively spliced exon 6A and update in mutation nomenclature". Human Mutation. 25 (5): 429–434. doi:10.1002/humu.20159. PMID 15832313. S2CID 12500736.
- Isaac, C.; Marsh, K. L.; Paznekas, W. A.; Dixon, J.; Dixon, M. J.; Jabs, E. W.; Meier, U. T. (September 2000). "Characterization of the nucleolar gene product, treacle, in Treacher Collins syndrome". Molecular Biology of the Cell. 11 (9): 3061–3071. doi:10.1091/mbc.11.9.3061. PMC 14975. PMID 10982400.
- Gorlin RJ, Syndromes of the Head and Neck, 2001, Oxford University Press, 4th edition
- Dixon, MJ; Marres, HA; Edwards, SJ; Dixon, J; Cremers, CW (April 1994). "Treacher Collins syndrome: correlation between clinical and genetic linkage studies". Clinical Dysmorphology. 3 (2): 96–103. doi:10.1097/00019605-199404000-00002. PMID 8055143.
- Dixon, Jill; Ellis, Ian; Bottani, Armand; Temple, Karen; Dixon, Michael James (15 June 2004). "Identification of mutations in TCOF1: Use of molecular analysis in the pre- and postnatal diagnosis of Treacher Collins syndrome". American Journal of Medical Genetics. 127A (3): 244–248. doi:10.1002/ajmg.a.30010. PMID 15150774. S2CID 2091796.
- Shoo, Brenda A.; McPherson, Elizabeth; Jabs, Ethylin Wang (1 April 2004). "Mosaicism of aTCOF1 mutation in an individual clinically unaffected with treacher collins syndrome". American Journal of Medical Genetics. 126A (1): 84–88. doi:10.1002/ajmg.a.20488. PMID 15039977. S2CID 35163245.
- Splendore, Alessandra; Jabs, Ethylin Wang; Félix, Têmis Maria; Passos-Bueno, Maria Rita (31 August 2003). "Parental origin of mutations in sporadic cases of Treacher Collins syndrome". European Journal of Human Genetics. 11 (9): 718–722. doi:10.1038/sj.ejhg.5201029. PMID 12939661.
- Senggen, E; Laswed, T; Meuwly, JY; Maestre, LA; Jaques, B; Meuli, R; Gudinchet, F (May 2011). "First and second branchial arch syndromes: multimodality approach" (PDF). Pediatric Radiology. 41 (5): 549–61. doi:10.1007/s00247-010-1831-3. PMID 20924574. S2CID 22416094. Archived (PDF) from the original on 2019-04-26. Retrieved 2020-01-15.
- Vento AR, et al., The O.M.E.N.S classification of hemifacial microsomia, 1991, Cleft Palate Craniofac, J 28, p. 68-76
- Posnick JC; et al. (2000). "Treacher Collins syndrome: current evaluation, treatment, and future directions". Cleft Palate Craniofac J. 55 (5): 1120–1133. doi:10.1597/1545-1569(2000)037<0434:TCSCET>2.0.CO;2. PMID 11034023.
- Dixon, MJ (1995). "Treacher Collins syndrome". Journal of Medical Genetics. 32 (10): 806–8. doi:10.1136/jmg.32.10.806. PMC 1051706. PMID 8558560.
- Goel L; et al. (2009). "Treacher Collins syndrome-a challenge for anaesthesiologists". Indian J Anaesth. 53 (4): 642–645. PMC 2894488. PMID 20640217.
- Evans, Adele Karen; Rahbar, Reza; Rogers, Gary F.; Mulliken, John B.; Volk, Mark S. (31 May 2006). "Robin sequence: A retrospective review of 115 patients". International Journal of Pediatric Otorhinolaryngology. 70 (6): 973–980. doi:10.1016/j.ijporl.2005.10.016. PMID 16443284.
- Rose, Edmund; Staats, Richard; Thissen, Ulrike; Otten, Jörg-Eland; Schmelzeisen, Rainer; Jonas, Irmtrud (1 August 2002). "Sleep-Related Obstructive Disordered Breathing in Cleft Palate Patients after Palatoplasty". Plastic and Reconstructive Surgery. 110 (2): 392–396. doi:10.1097/00006534-200208000-00002. PMID 12142649. S2CID 36499038.
- Bannink, Natalja; Mathijssen, Irene M. J.; Joosten, Koen F. M. (1 September 2010). "Use of Ambulatory Polysomnography in Children With Syndromic Craniosynostosis". Journal of Craniofacial Surgery. 21 (5): 1365–1368. doi:10.1097/SCS.0b013e3181ec69a5. PMID 20856022. S2CID 43739792.
- Marres, HA (2002). Hearing loss in the Treacher-Collins syndrome. Advances in Oto-rhino-laryngology. Vol. 61. pp. 209–15. doi:10.1159/000066811. ISBN 978-3-8055-7449-5. PMID 12408086.
- Zhang, Zhiyong; Niu, Feng; Tang, Xiaojun; Yu, Bing; Liu, Jianfeng; Gui, Lai (1 September 2009). "Staged Reconstruction for Adult Complete Treacher Collins Syndrome". Journal of Craniofacial Surgery. 20 (5): 1433–1438. doi:10.1097/SCS.0b013e3181af21f9. PMID 19816274. S2CID 44847925.
- Saadeh, Pierre B.; Chang, Christopher C.; Warren, Stephen M.; Reavey, Patrick; McCarthy, Joseph G.; Siebert, John W. (1 June 2008). "Microsurgical Correction of Facial Contour Deformities in Patients with Craniofacial Malformations: A 15-Year Experience". Plastic and Reconstructive Surgery. 121 (6): 368e–378e. doi:10.1097/PRS.0b013e3181707194. PMID 18520863. S2CID 27971712.
- Argenta, Louis C.; Iacobucci, John J. (30 June 1989). "Treacher Collins Syndrome: Present concepts of the disorder and their surgical correction". World Journal of Surgery. 13 (4): 401–409. doi:10.1007/BF01660753. PMID 2773500. S2CID 27094477.
- Marres, HA; Cremers, CW; Marres, EH (1995). "Treacher-Collins syndrome. Management of major and minor anomalies of the ear". Revue de Laryngologie – Otologie – Rhinologie. 116 (2): 105–108. PMID 7569369.
- Aug 12, Jane Ridley; 2022; Am, 10:56. "Man's parents rejected him at birth because of his appearance". Insider. Retrieved 2022-08-13.
{{cite web}}: CS1 maint: numeric names: authors list (link) - Conte, Chiara; Maria Rosaria D'Apice; Fabrizio Rinaldi; Stefano Gambardella; Federica Sanguiuolo; Giuseppe Novelli (27 September 2011). "Novel mutations of TCOF1 gene in European patients with treacher Collins syndrome". Medical Genetics. 12: 125. doi:10.1186/1471-2350-12-125. PMC 3199234. PMID 21951868.
- Treacher Collin E (1900). "Cases with symmetrical congenital notches in the outer part of each lid and defective development of the malar bones". Trans Ophthalmol Soc UK. 20: 190–192.
- Franceschetti A, Klein D (1949). "Mandibulo-facial dysostosis: new hereditary syndrome". Acta Ophthalmol. 27: 143–224.
- "Surgical Teamwork Gives Disease Victims a New Life" Archived 2018-07-23 at the Wayback Machine, Donald G. McNeil, Jr., July 26, 1977, page L31.
- "Bob Saget Biography". Retrieved 2022-06-30.
{{cite web}}: CS1 maint: url-status (link) - "Nip/Tuck: Blu Mondae - TV.com". Archived from the original on 2008-06-12. Retrieved 2008-05-12.
- "First Coast News: Local Family Has Daughter Born Without a Face".
- "BBC programme page for Love Me, Love My Face". BBC Three. 17 June 2011. Archived from the original on 21 January 2018. Retrieved 7 November 2017.
- "BBC programme page for So What If My Baby..." BBC Three. 24 August 2011. Archived from the original on 2 June 2017. Retrieved 7 November 2017.
- "BBC Three Bringing Up Britain season". BBC One. 12 April 2011. Archived from the original on 12 August 2011. Retrieved 24 August 2011.
- "Finding My Family on Facebook". bbc.co.uk. BBC Three. 2011. Archived from the original on 2011-10-31..
- Chilton, Martin (24 February 2012). "Wonder by R. J. Palacio: review". The Telegraph. Archived from the original on 6 October 2017. Retrieved 7 November 2017.
- "Julia Roberts' Drama 'Wonder' Pushed to November". The Hollywood Reporter. February 13, 2017. Archived from the original on April 22, 2017. Retrieved February 13, 2017.
- O'Conner, Katie (December 22, 2017). "Real Life Reflected on the Silver Screen". Richmond Times-Dispatch.
- Wilner, Norman (February 18, 2020). "Canadian Screen Awards 2020: Prepare for a Schitt's show". Now. Archived from the original on 2020-02-18. Retrieved December 28, 2020.